DOI:10.32604/biocell.2022.017248
| BIOCELL DOI:10.32604/biocell.2022.017248 | |
| Review |
Mesenchymal stem cells: As a multi-target cell therapy for clearing β-amyloid deposition in Alzheimer’s disease
1Laboratory Animal Center, Shanxi Medical University, Taiyuan, 030000, China
2Department of Orthopaedics, Second Hospital Shanxi Medical University, Taiyuan, 030000, China
*Address correspondence to: Chunfang Wang, wangchunfang@sxmu.edu.cn
Received: 26 April 2021; Accepted: 21 June 2021
Abstract: Extracellular β-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs) are the pathological hallmarks of Alzheimer’s disease (AD). Studies have shown that aggregates of extracellular Aβ can induce neuroinflammation mediated neurotoxic signaling through microglial activation and release of pro-inflammatory factors. Thus, modulation of Aβ might be a potential therapeutic strategy for modifying disease progression. Recently, a large number of reports have confirmed the beneficial effects of mesenchymal stem cells (MSCs) on AD. It is believed to reduce neuroinflammation, reduce Aβ amyloid deposits and NFTs, increase acetylcholine levels, promote neurogenesis, reduce neuronal damage, and improve working memory and cognition. In this review, we focus on the role of MSCs in clearing Aβ deposition. MSCs have the potential to modulate Aβ-related microenvironments via enhancement of autophagy, proteolysis of Aβ aggregates, phagocytic clearance of Aβ by microglial M2 polarization, decrease oxidative stress (OS), and correction of abnormal sphingolipid (SL) metabolism. With advantages in clinical applications, these data suggest that the use of MSCs as a multi-target modulator of Aβ would be an effective therapeutic approach in AD.
Keywords: Alzheimer’s disease; β-amyloid; Microglia; Neurodegeneration; Stem cell therapy
Alzheimer’s disease (AD) is the most common chronic neurodegenerative disease, with increased life expectancy, this number is expected to rise in the future (Moonga and Likupe, 2016). AD is characterized by progressive memory deficits, cognitive impairment, and personality changes associated with the degeneration of multiple neuronal types and pathologically by synapse loss and the presence of β-amyloid (Aβ) plaques and NFTs (Karlawish et al., 2005). All types of AD are characterized by the accumulation of Aβ, which triggers a series of neuroinflammation, culminating in neuronal dysfunction and death. Aβ plays a critical pathogenic role in AD and is related to the enhancement of oxidative stress (OS) in the brain, neuronal damage, synapse loss, and NFTs (Cunningham, 2013; Götz et al., 2001).
Cell therapy has been regarded as one of the most promising novel therapies as a disease-modifying strategy for AD (Si et al., 2011). Mesenchymal stem cells (MSCs), as a kind of stem cell that is easily available, has low immunogenicity, strong proliferation, and differentiation potential, have been used in the research of various diseases. Currently, we can isolate and prepare MSCs from bone marrow, fat, synovium, bone, muscle, lung, liver, pancreas, amniotic fluid, and umbilical cord blood. By using cell replacement or immunomodulation strategies, isolated MSCs have a beneficial effect on neurodegenerative diseases (including AD) in animal models (Sadan et al., 2012; Gugliandolo et al., 2019; Chen et al., 2020b; Kim et al., 2020; Reyhani et al., 2020). Initially, people considered that it could replace the death and loss of AD neurons by differentiation into neurons (Mezey et al., 2003). Since then, it has been suggested that in neuron replacement therapy, there are still several obstacles to be resolved, such as the transplantation pathway, tumorigenesis and mutation, and the efficiency of neuronal differentiation (Prockop et al., 2003; Caruso and Parolini, 2015). However, with the in-depth study of stem cells, more and more evidence has supported that MSCs enter the blood-brain barrier (BBB) by paracrine with soluble factors to improve the brain microenvironment (Ma et al., 2013; Lim et al., 2020; Chen et al., 2020a; Mehrabadi et al., 2020).
The neuroprotective mechanism, by which MSCs indirectly modulate Aβ-related microenvironments, appears to be complex and pleiotropic. In this review, we provide evidence that MSCs clear Aβ via enhancement of autophagy-related proteins, autophagy-lysosome metabolism, and protein degrading enzyme levels, decrease of neuroinflammation, and controlling microglial polarization. We also discussed the implications of these findings for therapeutic interventions and the prospects for MSCs in the treatment of AD.
Structure and formation of β-amyloid
As we all know, Aβ is a hydrolysate of amyloid precursor protein (APP), and APP is continuously cleaved by β-/γ-secretase to form amyloid proteins (Masters and Selkoe, 2012). APP is hydrolyzed by β-secretase (also known as β-site amyloid cleavage enzyme, BACE) at the first amino acid position of the Aβ sequence to produce a large N-terminal fragment (sAPP β) and a small transmembrane fragment (C99), the latter acts on the 40/42 amino acid position of the Aβ sequence by γ secretase to produce a 39-42 amino acid peptide-Aβ and another intracellular fragment APP intracellular domain (AICD) (Caruso and Parolini, 2015; Nikolaev et al., 2009). Current studies have shown that the aggregation activity and toxicity of Aβ42 are the main pathogenic factors that promote the development of AD.
Part of the causes of β-amyloid deposits
Microglia-associated immune response
Under normal physiological conditions, neurons and glial cells in the central nervous system together maintain the homeostasis of the central nervous system. The stability of the content of Aβ in the brain depends on the balance between Aβ production and clearance, and the clearance of Aβ largely depends on the phagocytosis of microglia. After being activated by Aβ, microglia can phagocytose Aβ aggregates and necrotic nerve cells (Moore et al., 2002; Rogers et al., 2002). The clearance of Aβ also depends on the amyloid degrading enzymes (ADE) secreted after activation of microglia, such as neutral endopeptidase (NEP) and insulin-degrading enzyme (IDE) (Farris et al., 2003; Miller et al., 2003; Kim et al., 2020). Therefore, in the early stage of AD, microglia can effectively delay the development of AD through phagocytosis and production of degrading enzymes. However, in the late stage of AD, the physiological function of microglia cells is impaired (Morgan, 2018), resulting in excessive deposition of Aβ. Currently, amyloid deposits can bind to receptor proteins on the surface of microglia, such as TLR receptors and sweeper receptors, and enter cells through endocytosis. But at the same time, microglia are also activated abnormally, releasing neuroinflammatory cytokines and inflammatory mediators, further exacerbating local inflammatory responses and causing irreversible damage to neurons (Hong and Stevens, 2016).
Microglia can recognize various pathogens and endogenous cues. These cues in turn produce morphological changes, usually described as the M1 or M2 phenotype of microglia. Studies have shown that Aβ aggregates released from neurons directly induce microglia to transfer to the more reactive M1 phenotype, leading to inflammatory cascades (Neniskyte et al., 2016; Boche et al., 2013). Further increase of pro-inflammatory factors, such as IL-1, NO, and TNF, leads to the formation of amyloid plaques and neurodegenerative changes. In contrast, M2 polarized microglia is induced by IL-4 and/or IL-10, are believed to have anti-inflammatory effects, can promote phagocytosis, and are known to express arginase-1 (Arg-1), MRC1, and YM-1 (Ma et al., 2013). Furthermore, “M2 microglia” is indicated to increase the expression of the two main Aβ-degrading enzymes (IDE and NEP) in the brain (Wang et al., 2010; Edbauer et al., 2002). A large amount of evidence from AD studies shows that M2 polarization is closely related to β-amyloid clearance (Ma et al., 2013; Rogers et al., 2002; Hong and Stevens, 2016; Mandrekar-Colucci and Landreth, 2010). Therefore, it is important to modify microglial function, as a treatment mechanism to induce the clearance of Aβ before the formation of plaques, thereby improving the pathology of AD.
Autophagy failure: a novel factor in β-amyloid deposition
Autophagy is the process of transporting damaged, aging, or incorrectly aggregated proteins and organelles to lysosomes for degradation, to maintain the homeostasis of the intracellular environment. In the past five years, more and more wonderful reviews have summarized autophagy as a target for the treatment of neurodegenerative diseases (Park et al., 2020b; Suresh et al., 2020; Fujikake et al., 2018). The downregulation of autophagy was morphologically and genetically demonstrated in AD brains (Xiao et al., 1999). Morphologically, a large number of autophagic vacuoles (AVs) and lysosomes have accumulated in the brain of patients with AD with dystrophic neuritis (Lee et al., 2011; Whyte et al., 2017). Presenilin 1 (PSEN1/PS-1) is a component of the secretase complex that generates peptides (Saftig et al., 1998). A recent study showed that PS-1 is essential for v-ATPase targeting to lysosomes, lysosome acidification, and proteolysis during autophagy. The mutation of PS-1 in familial AD results in abnormal transport of v-ATP enzyme to lysosomes, which leads to lysosomal alkalization and autophagy-lysosome accumulation (Coffey et al., 2014; Lee et al., 2010c).
In general, studies have shown that rats lacking the Atg5/Atg7 gene have the AD phenotype (Hara et al., 2006). Furthermore, Beclin1, a key regulatory protein of autophagy, was identified as a causative molecule in AD pathology (Hara et al., 2006; Rocchi et al., 2017; Swaminathan et al., 2016). Vps34 is a mammalian class III PI3K enzyme, which mainly plays a role in the early stages of autophagy. Vps34 combines with Beclin1 in a variety of enzymatic reactions to form a Vps34/PI3K-Beclin1 complex, which participates in the maturation and transport of autophagic vesicles (AVs) and promotes the formation of autophagosomes under the action of Dapper1 regulatory factors (Ma et al., 2014). In AD, the complex is abnormal due to the decrease of Beclin1. This will lead to inhibition of the formation and maturation of autophagy vesicles and further reduce the occurrence of autophagy. This will lead to a decrease in Aβ clearance and induce AD (Jaeger et al., 2010) (Fig. 1). These studies indicate that Beclin1 plays a central role in the progression of AD and makes Beclin1 a target for improving Aβ starch deposition.
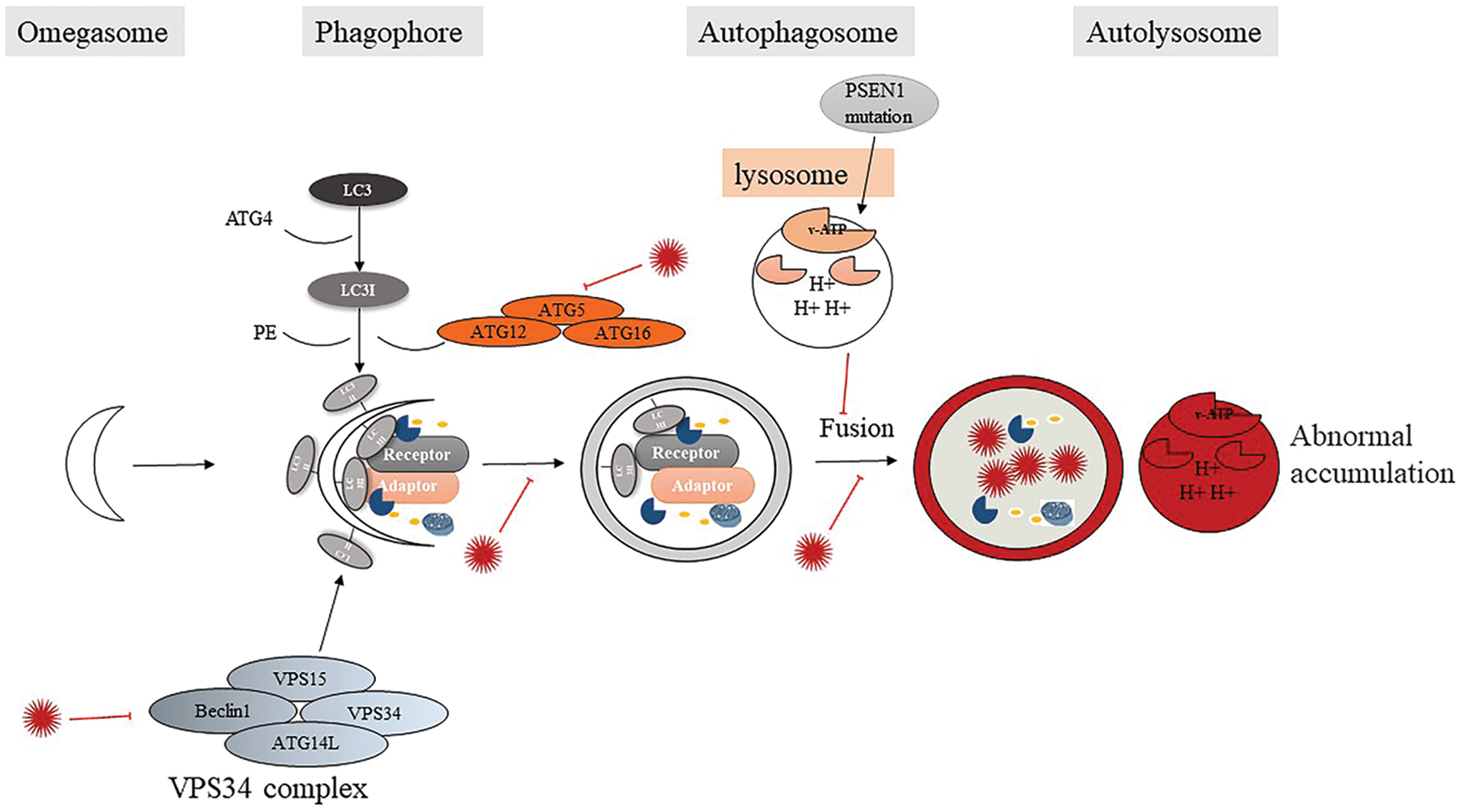
Figure 1: Autophagy failure: a novel factor in β-amyloid deposition.
Interactions between OS and β-amyloid
The aggregation of Aβ amyloid is related to the increase of OS. On the one hand, the increase of OS may be related to mitochondrial dysfunction and lipid peroxidation caused by amyloid aggregation (de la Monte and Wands, 2006). The aggregation of soluble Aβ induces reactive oxygen species (ROS), leading to synaptic damage and neuron loss in hippocampal neurons (de Felice et al., 2007). These studies indicate that Aβ amyloid deposition contributes to the increase of OS in AD models.
On the other hand, the increase of OS in the brain further causes the accumulation of Aβ. The neurotoxicity of Aβ comes from the methionine residues of the peptide carbon segment, which can generate ROS, induce oxidation reactions, and produce peroxides, which can further cause the Aβ soluble body to transform into Aβ insoluble body, and finally constitute age spots. A study showed that OS can lead to an increase in Aβ levels in cultured neuroblastoma cells (Misonou et al., 2000); it was reported that the loss of antioxidant function in Tg19959 mice overexpressing double-mutated APP caused an increase in OS and significantly increased Aβ levels and Aβ plaque deposition in the brain (Li et al., 2004). Studies have shown that in differentiated human neuroblastoma cells, dual regulation of amyloid precursor protein metabolism is associated with downregulation of α-secretase and upregulation of γ-secretase, particularly β-secretase and JNK-dependent a β generation (Quiroz-Baez et al., 2009). Since OS-mediated neurotoxicity and Aβ amyloid deposition are key factors leading to neurodegenerative diseases, the development of effective antioxidant protection is an attractive strategy for AD treatment.
Sphingolipid (SL) metabolism involved in β-amyloid
Previous studies have reported that abnormal SL patterns lead to SL-protein interactions, which to some extent lead to misfolding events, such as amyloid deposition in AD (Piccinini et al., 2010; Matsuzaki, 2020). It is reported that brain glycosphingolipids (GSL) metabolism disorders have appeared in the brains of patients with AD and transgenic mouse models of the disease (Ariga et al., 2008). GSL participates in the formation of β-amyloid in these ways: on the one hand, it regulates the functions of APP as a signal molecule and the proteolytic process of APP in the process of amyloidosis; on the other hand, it promotes the conversion of soluble β-starch into insoluble form (Piccinini et al., 2010). Studies have analyzed several SL and SL hydrolases in brain samples of AD patients and age-matched normal individuals. The pattern of increased expression of acid sphingomyelinase (ASM) and acid ceramidase in AD was found, resulting in a decrease in sphingomyelin and an increase in ceramide. The downstream result of ASM activation is increased ceramide, activation of ceramidase, and sphingosine production. The decrease in sphingosine-1-phosphate (S1P) levels in the brain of patients with AD and the increase in ceramide levels may contribute to the pathogenesis of the disease (He et al., 2010).
MSCs are known for their strong proliferation, multi-lineage differentiation, immune regulation, and a wide range of sources. They include bone marrow MSCs (BM-MSCs) (Lee et al., 2010b), adipose-derived MSCs (AD-MSCs) (Ma et al., 2013), umbilical cord MSCs (hUCMSCs) (Ding et al., 2018), placental MSC and so on. They can differentiate into non-mesoderm cell types (including neuronal lineages) in vivo and in vitro (Li et al., 2015). MSCs have strong immune regulation and regeneration capabilities, which stem from their ability to secrete a variety of chemokines, cytokines, and nutritional factors, and have been shown to have a regulatory effect on the progression of AD under many different conditions (Ma et al., 2013; Kim et al., 2018a; Lee et al., 2012b). The most important thing is that these small molecules can enter the brain through the BBB to regulate the microenvironment in the BBB (Wang et al., 2018). At least it can be confirmed that MSCs can target the removal of Aβ protein deposits through the following aspects.
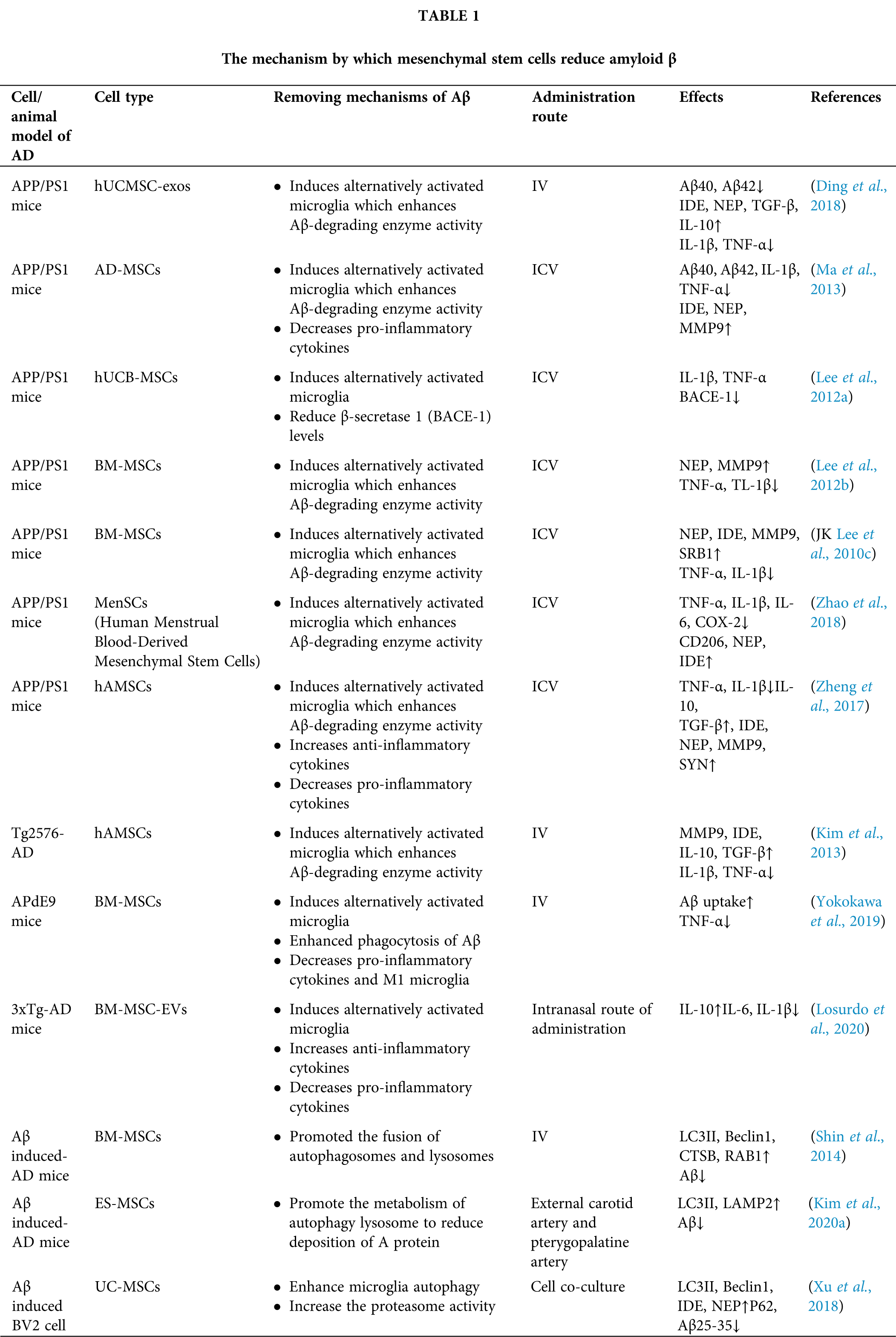
MSCs regulate β-amyloid degradation through a variety of pathways (Table 1)
MSCs reduce Aβ and neuroinflammation via microglia activation
Microglia is an immune regulatory cell of the nervous system, which initiates an immune response and induce neuroinflammation in the AD brains through toll-like receptors (TLR2, TLR4, TLR6, and TLR9) (Heneka et al., 2015b; Heneka et al., 2015a; Lee et al., 2009). However, ATSCs-conditioned medium can reduce the excessive activation of microglia and reduce the secretion of pro-inflammatory factors IL-1β and TNF-α by reducing the activation of TLR2 and TLR4 receptors in microglia (Mehrabadi et al., 2020). Lee et al. (2010b) implanted MSCs into the brains of APP/PS1 transgenic mice and found that the phenotype of microglia in the cerebral cortex and hippocampus of mice changed to M2 type (Lee et al., 2010b). This effect was also demonstrated in the acutely induced AD mice model, BM-MSCs can prevent and/or eliminate Aβ deposition.
In addition, MSCs can also eliminate Aβ by enhancing the secretion of Aβ proteolytic enzymes in microglia or upregulating the expression of uptake Aβ receptors in activated microglia (Lee et al., 2010b; Kim et al., 2013; Yokokawa et al., 2019; Kim et al., 2018b; Zheng et al., 2017; Zhao et al., 2018; Lee et al., 2009). On the one hand, M2 microglia enhanced the secretion of Aβ proteolytic enzymes NEP, IDE, and matrix metallopeptidase 9 (MMP9). On the other hand, it up-regulated the expression of Aβ protein binding receptor scavenger receptor B-1 (SCARB-1) and enhanced the uptake of Aβ amyloid (Kim et al., 2013). Yokokawa et al. (2019) also confirmed in APdE9 transgenic mice that BM-MSCs can change the phenotype of microglia by up-regulating CD14, enhancing the uptake of Aβ and inhibiting the production of pro-inflammatory factors (Yokokawa et al., 2019). And an in vivo experiment showed that Human umbilical cord blood-derived MSCs (hUCB-MSCs) treatment can significantly reduce the level of BACE-1 (Lee et al., 2012a). MSCs regulate the function of microglia in AD by activating neuroprotective microglia (Fig. 2).
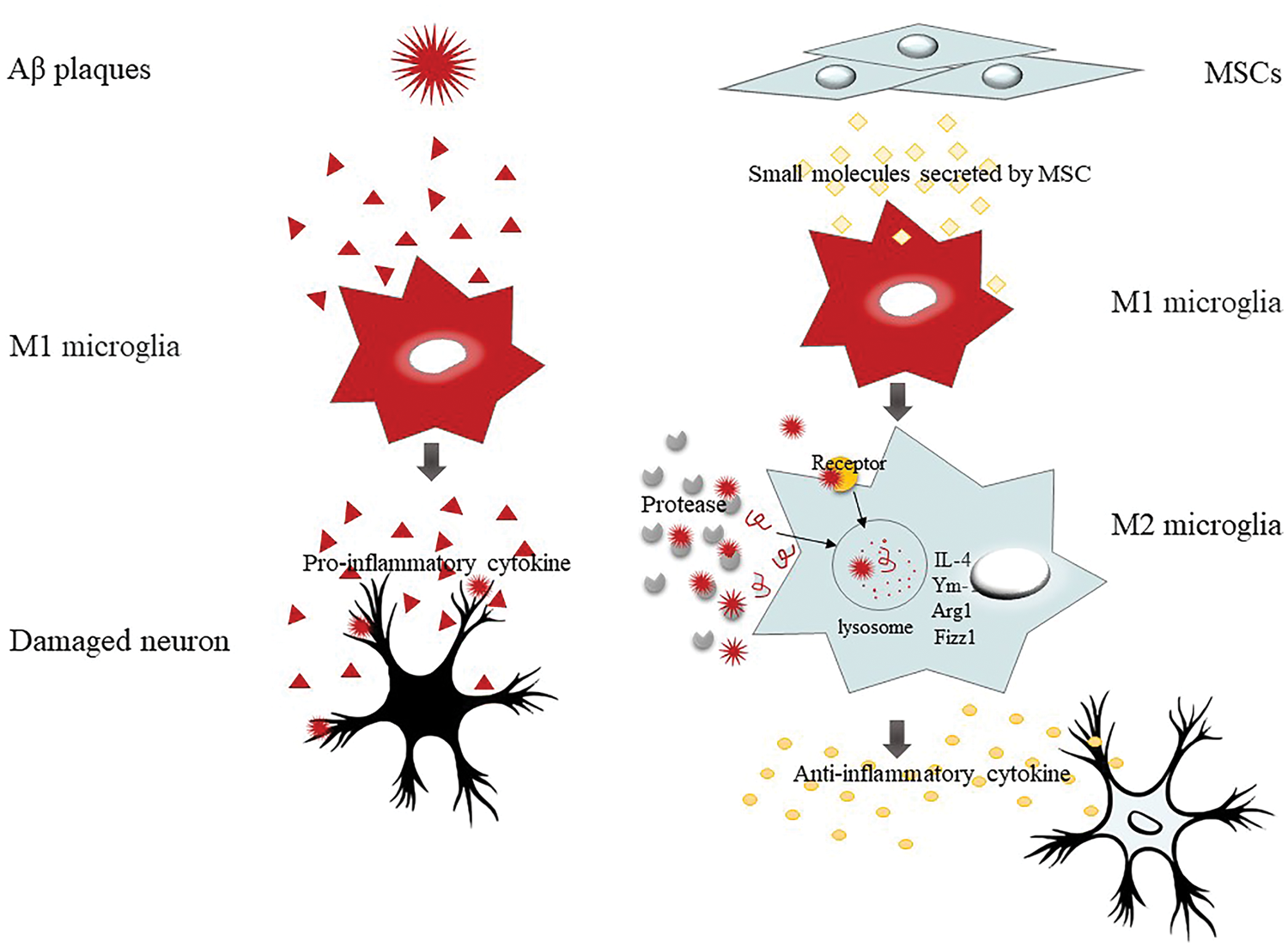
Figure 2: MSCs have immunoregulatory properties, especially microglia polarization.
With the deepening of research on MSCs, more and more people are interested in the mechanism by which MSCs function. Due to its limited efficiency of directional differentiation into damaged neurons in AD models (Parr et al., 2007), more people place their hopes on its paracrine mechanism or transplantation after directional differentiation in vitro (Ma et al., 2013; Yokokawa et al., 2019; Ding et al., 2018; Losurdo et al., 2020; Kim et al., 2018b; Lee et al., 2012b). Recent studies have demonstrated the immunomodulatory effect of MSCs-derived extracellular vesicles in different AD models (Ding et al., 2018; Losurdo et al., 2020). hUCMSCs-derived exosomes (hUCMSCs-Exo) improves the microenvironment by inducing the transformation of microglia into a neuroprotective phenotype, and by up-regulating NEP and IDE to reduce Aβ deposition and enhance the secretion of neuroprotective factors in microglia (Ding et al., 2018). In addition, in the 3xTgAD model, nasal transplantation of BM-MSC-EV exerts an immunomodulatory effect promotes microglia to polarize towards an anti-inflammatory phenotype and improves synaptic loss (Losurdo et al., 2020). However, after Yokokawa et al. (2019) filtered the BM-MSC culture medium with a 100 kDa ultrafilter to filter out the cell vesicles, the effect of activating microglia remained (Yokokawa et al., 2019). After further filtering out more molecules with finer filtering conditions, this effect disappeared. The data showed that in addition to extracellular vesicles, the secretome secreted by MSCs also enhanced the uptake of Aβ deposition by microglia.
A recent report indicated that CCL-5 secreted by BM-MSCs can recruit bone marrow-derived microglia and induce the immune response of microglia to improve the neuropathology of AD.CCL-5, which can control the migration of T lymphocytes, mononuclear macrophages, and eosinophils (Ransohoff et al., 2007), are widespread in neurodegenerative diseases (Mines et al., 2007). Existing evidence suggests that this chemokine can promote the migration of M2 microglia instead of M1 in spinal cord injury. In the study of Jong Pil Lee, in vitro experiments found that when the microenvironment changes of the AD brain were simulated, the migration of microglia increased significantly after BM-MSCs treatment, and the level of CCL-5 also increased significantly (Lee et al., 2012b). Secondly, in APP/PS1 animal experiments, it was found that endogenous microglia can clear Aβ by enhancing the secretion of Aβ degrading enzymes (including NEP and MMP). Interestingly, these effects will disappear after transfecting BM-MSCs with CCL-5 siRNA (Lee et al., 2012b). In addition, Dong Hyun Kim verified the effect of GDF-15 secreted by hUCB-MSCs on Aβ amyloid in 5XFAD mice and in vitro models. GDF-15 is a therapeutic neurotrophic factor. In AD, it can promote the generation of hippocampal nerves and synaptic activity. The loss of GDF in neonatal mice is manifested as progressive loss of motor and sensory neurons (Kim et al., 2018a). TGFβRII is a receptor involved in the GDF-15 pathway, which is related to the mechanism of clearing Aβ42. In this study, GDF-15 secreted by hUCB-MSCs mainly corrected the expression of TGFβRII receptor and the degrading enzyme IDE in microglia. After knocking out GDF-15 in hUCB-MSCs, these effects disappeared (Kim et al., 2018a). These data indicate that MSCs can increase, through paracrine action, the ability of microglial cells to clear Aβ.
MSCs reduce Aβ and neuroinflammation via modulating autophagy
Research evidence shows that autophagy plays a key role in AD, and the disorder of autophagy function may lead to the accumulation of Aβ amyloid. Currently, researchers believe that the role of autophagy in MSCs includes two aspects: ① MSCs can regulate cell autophagy involved in diseases or regulate the expression of autophagy-related proteins to treat diseases (Fig. 3); ② Autophagy is maintained MSCs play a critical role in many aspects such as secretion, proliferation, migration, and differentiation (Fig. 4).
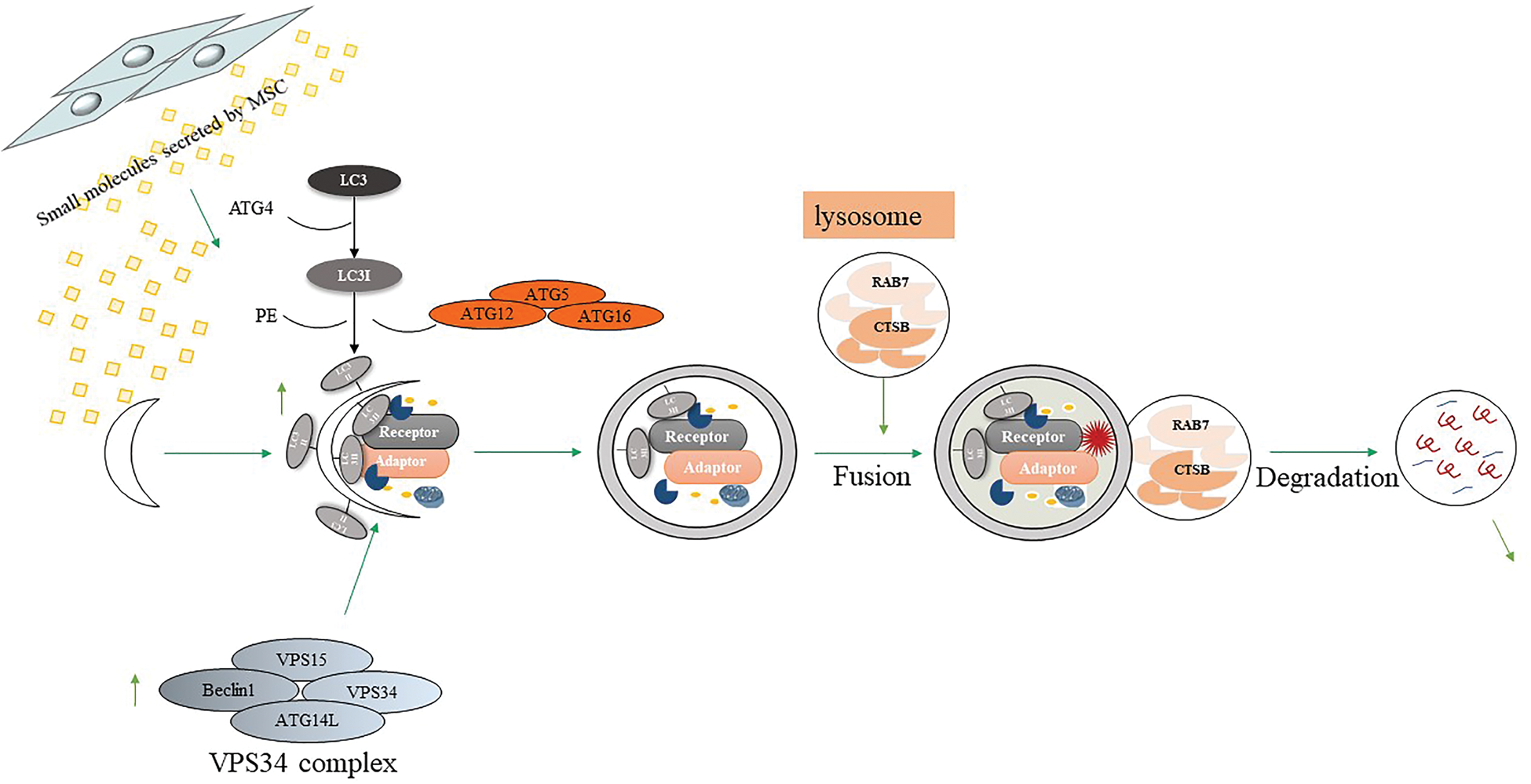
Figure 3: MSCs-derived small molecules enhance Aβ degradation by autophagy.
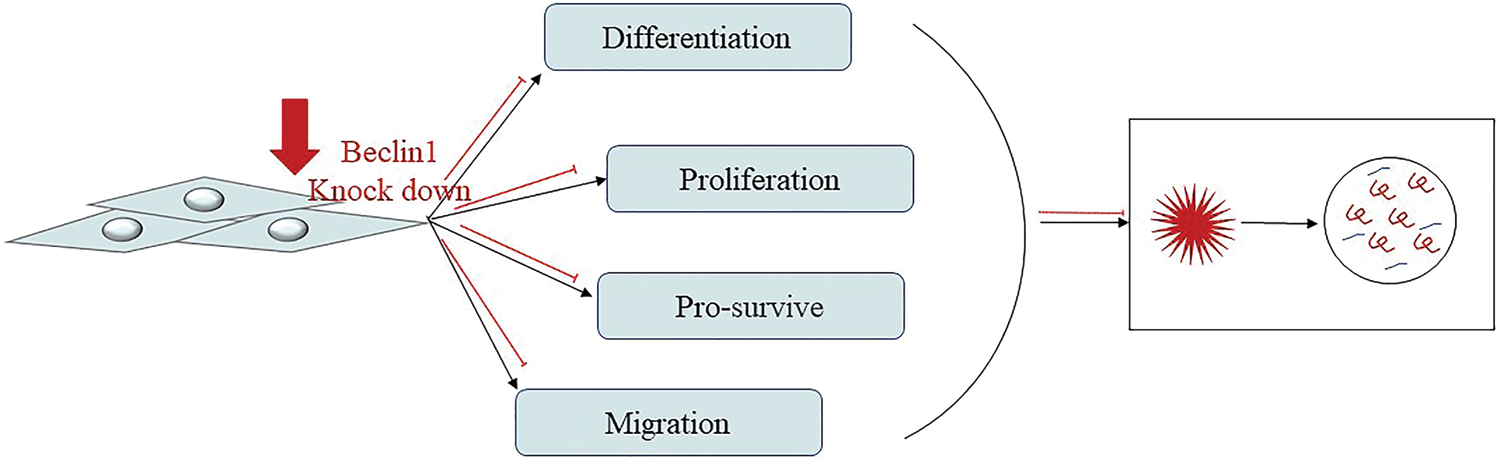
Figure 4: MSCs require autophagy to maintain their ability to proliferate, migrate, differentiate, and survive.
According to a report, in SH-SY5Y cells treated with Aβ, autophagolysosomal increased, and most autophagosomes did not fuse with lysosomes, which is an important cause of neuronal damage. However, after MSCs were co-cultured with SH-SY5Y cells treated with Aβ, the expression of LC3II and the major protease cathepsin B (CTSB) in the lysosomes increased, indicating an increase in the formation of autophagosomes. In addition, MSCs intervention also increased the expression of RAB7 (which is necessary for the final maturation of late AVs and fusion with lysosomes), proving that MSCs promoted the fusion of autophagosomes and lysosomes (Shin et al., 2014). This means that under the treatment of Aβ, MSCs can significantly increase the formation of neuronal autophagosomes and promote the metabolism of lysosomes. In the Aβ-treated mice, the autophagy regulation effect of MSCs is more significant, mainly in inducing the formation of autophagosomes, promoting the maturation of AVs and fusion with lysosomes, and further down-regulating Aβ-treated mice Medium Aβ level, increase the survival rate of neurons. In addition, MSCs treatment can up-regulate the expression of Beclin1 in vivo and in vitro experiments (Shin et al., 2014). Moreover, the efficacy of autophagy induction in ES-MSCs was comparable to that of BM-MSCs (Kim et al., 2020a). A recent study proved that cytokines secreted by MSCs can enhance the expression of autophagy-related proteins in microglia. Umbilical cord mesenchymal stem cell-conditioned medium (ucMSCs-CM) was co-cultured with BV2 microglia (Xu et al., 2018). It was found that ucMSCs-CM can increase Aβ25-35 phagocytosis by enhancing the autophagy of microglia compared with the control group without MSCs medium and changes the expression of autophagy-related proteins. Taken together, these findings suggest that, although the exact pathological role of autophagy in AD remains to be elucidated, MSCs can serve as autophagy inducer that may provide new effective therapeutic strategies through degrading Aβ in the early AD.
On the contrary, some scientists believe that MSCs need autophagy to maintain their function and improve cognitive impairment in APP/PS1 mice. In order to study the role of autophagy in hUCMSCs, Li et al. (2018) knocked out Beclin1 in cells and found that in addition to changes in autophagy-related proteins (LC3, Beclin1, ATG7, and P62), migration and differentiation (SDF-1), stem cell key protein (Sox2), and apoptosis (caspase-3 and PARP) related proteins have also changed. It is proved that autophagy plays a vital role in maintaining the differentiation, migration, and survival of MSCs (Fig. 4). In addition, hUCMSCs transplantation can also improve the working memory and LTP of AD mice, while significantly inhibiting DEP, an indicator of reverse learning behavior. In the hippocampus and cerebral cortex, it reduces apoptosis and increases the new-born neuron. However, after inhibiting the autophagy of hUCMSCs, these effects all disappeared (Li et al., 2018). A recent study showed that the regulation of autophagy may also affect the secretion capacity of BM-MSCs, thereby affecting their functions. In fact, subcutaneous injection of BM-MSCs pretreated with the autophagy inducer rapamycin can enhance the wound healing ability of BM-MSCs. On the contrary, the therapeutic effect of early Beclin1 silencing BM-MSCs was reduced (An et al., 2018). These findings demonstrate that autophagy plays a key role in the treatment of MSCs, but now, more convincing evidence is still needed and still represents a stimulating field of research.
OS is considered to be an important factor of neurotoxicity. It occurs in the early stage of AD. Before the appearance of clinical and pathological symptoms, it is considered that it may be one of the causes of Aβ starch deposition (Jiao et al., 2016). Previously, studies have confirmed that MSC can induce antioxidant effects in neurodegenerative diseases (Yokokawa et al., 2019; Godoy et al., 2018; Wang et al., 2018). Godoy and his colleagues proposed that MSC-EVs exerts a neuroprotective effect due to the presence of antioxidant enzymes, anti-inflammatory, and/or nutritional molecules. Studies have shown that MSC-EVs contains and carries catalase, which makes EVs have ROS scavenging activity (Godoy et al., 2018).
Glutathione (GSH) is a critical cellular antioxidant stressor, which may reduce from oxidation by hydrogen peroxide and hydroperoxides, and it protects the protein thiol (Behl, 2005). Glutathione is converted to GSSG through GPx, to detoxify peroxide, glutathione reductase can reduce the reaction. GSH/GSSG is an index to evaluate the anti-oxidation in the cell. SOD is an important antioxidant enzyme that catalyzes the generation of H2O2 from superoxide radical anions and induces the oxidation of polyunsaturated fatty acids and lipid peroxidation (Smith et al., 2000). MDA is the final product of lipid peroxidation and has a toxic effect on neurons. A recent study showed that the tail vein injection of human amniotic MSCs (hAMSCs) can alleviate OS by reducing lipid peroxide products and increasing the levels of antioxidant enzymes SOD and GSH (Jiao et al., 2016). Compared with the PBS control group, hAMSCs treatment significantly increased the SOD activity in the brain of AD transgenic mice and reduced the MDA level.
Furthermore, in the normal brain, Selective Alzheimer’s disease indicator-1 (seladin-1) protects neurons from OS (Greeve et al., 2000). When OS in AD increases, seladin-1 is cleaved in a caspase-dependent manner (caspase 6 and caspase 3) and may be inactivated (Greeve et al., 2000). Mo et al. reported that BM-MSCs significantly down-regulate the expression and activity of caspase-3, thereby protecting seladin-1 from lysis (Mo et al., 2012). Research by Salem showed that BM-MSCs can repair damaged brains and significantly increase the levels of choline acetyltransferase (ChAT) and survivin expressing cells (Salem et al., 2015). And it up-regulates the expression of selective seladin-1, Nestin, and further removes β amyloid plaques in the hippocampus And MSCs can improve biomarkers better than drugs (Salem et al., 2015). In addition, studies have reported that in vitro induction of hMSCs to neurons (hMSCs-N) is more resistant to Aβ42 aggregates than undifferentiated MSCs (Cecchi et al., 2011). hMSCs-N enhanced its resistance to Aβ toxicity by reducing the levels of membrane GM1, Ca2+, and ROS in neurons (Cecchi et al., 2011). These results indicate that MSCs as effective antioxidants reduce Aβ deposition better than drugs.
MSCs have the potential to correct SL metabolism
As AD progresses, abnormal SL metabolism is often observed. It is related to dysfunctional protein clearance, impaired lysosome production, and inflammation control functions, as well as Aβ production and autophagy (Takasugi et al., 2011; Maceyka et al., 2012; Czubowicz et al., 2019). These experiments investigated the possibility of using stem cells to treat abnormal SL metabolism (Lee et al., 2010a; Marfia et al., 2016). The BM-MSCs were transplanted into the cerebellum of the Niemann-Pick type C disease mouse model, and the correction of S1P levels and the decrease of sphingosine accumulation were observed. Therefore, it can reduce cell apoptosis, restore calcium homeostasis, and prevent neuron loss (Lee et al., 2010a). In another study, the conditioned medium of AD-MSCs controlled the sphingomyelin kinase/sphingomyelin-1-phosphate signaling pathway and inhibited the activation of microglia (Marfia et al., 2016). These studies demonstrate that MSCs have the potential to correct SL metabolism abnormalities in mice. Although no one has yet studied the effect of MSCs on abnormal SL metabolism in AD brains, we believe that this is an area worthy of further study.
Conclusion and Future Perspective
As a promising method of treatment, MSCs therapies have become at the forefront of the field of neurodegeneration disease, especially in the area of AD. It is believed to reduce neuroinflammation, clear Aβ deposits, neurofibrillary tangles, increase acetylcholine levels, promote neurogenesis, reduce neuronal damage, improve working memory and cognition (Ma et al., 2013; Ma et al., 2020; Wang et al., 2018). In almost all types of ADs, Aβ plays a key pathogenic role, and studies have connected Aβ plaques with the formation of intercellular tau tangles and neuroinflammation. Therefore, we focus on the degradation effect and mechanism of Aβ by MSCs in this review. Considering that the number of MSCs differentiated into neuronal cells after transplantation in vivo is very small (Prockop et al., 2003; Caruso and Parolini, 2015), the cell-free therapy using MSCs-derived secretome might constitute an alternative because of their advantages. Many studies suggest that MSC-secretomes act as an important mediator of the information exchange between MSCs and neurons in neurodegenerative diseases (Kim et al., 2010; Lim et al., 2020; Wang et al., 2018). Specifically, MSC-secretomes contain neuroregulatory molecules known as potential therapeutic mediators against AD-related microenvironments. Recent studies have shown that clearance mechanisms of MSC-secretomes or exosomes in Aβ are likely multifaceted, including immune regulation, increased Aβ-degrading factors, enhanced autophagy, reduced OS, and correction of SL metabolism, which suggests the potential of clinical application for the treatment of AD (Fig. 5) (Yokokawa et al., 2019; Kim et al., 2018a; Shin et al., 2014; Kim et al., 2013; Park et al., 2020a; Lee et al., 2010a; Wang et al., 2018; Marfia et al., 2016; Jiao et al., 2016; Kim et al., 2020a).
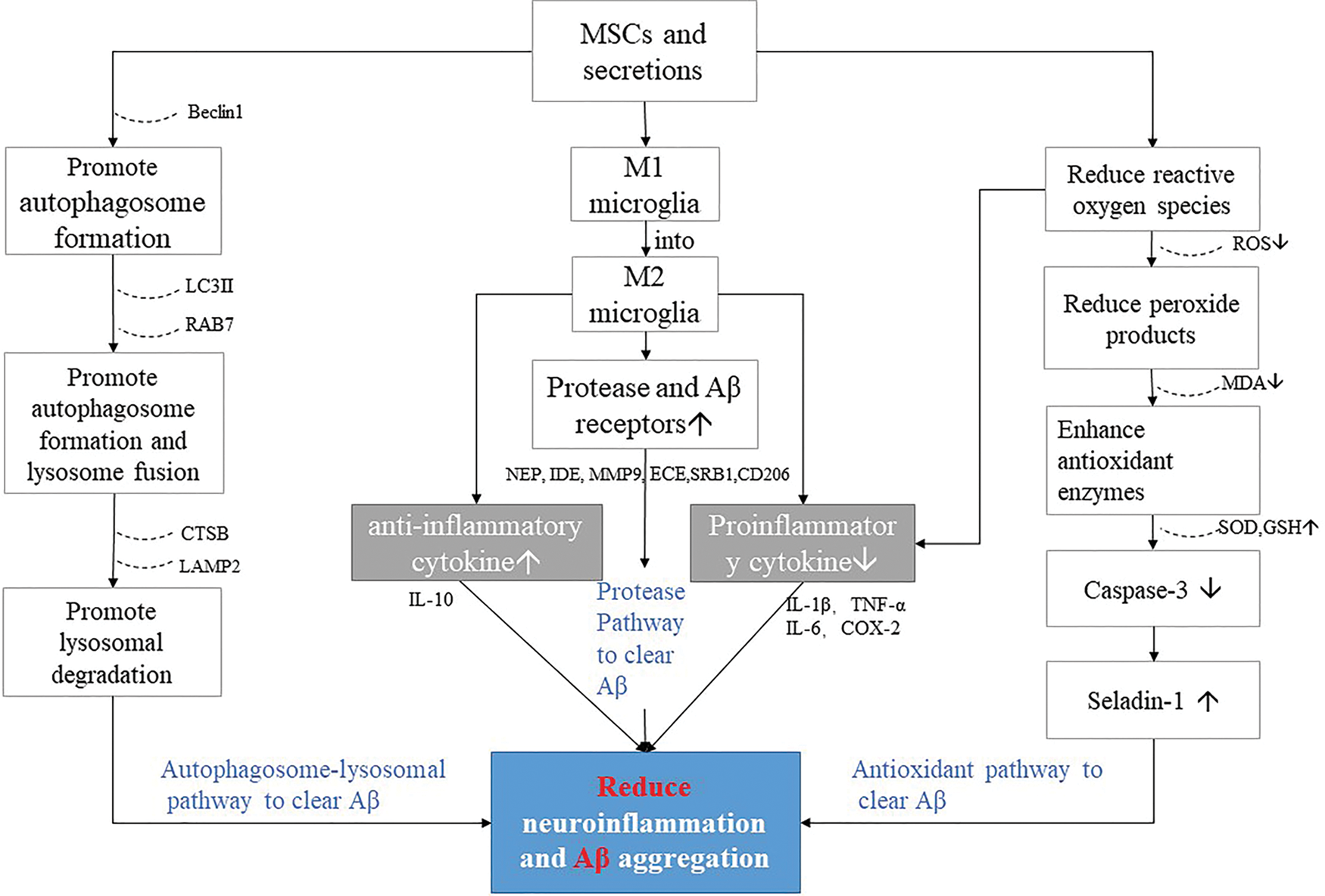
Figure 5: MSCs and secretions factors reduce pathways of neuroinflammation and Aβ aggregation in AD.
The accumulation of Aβ is the result of many factors, so its clearance should also be multi-targeted. The fact that a huge amount of money has been put into the development of protein-based therapies in AD but has not achieved valid results supports this issue in part (Salloway et al., 2014). Therefore, a strategy that can modulate multiple pathological factors of AD is important to achieve clinical benefits in the treatment of neurodegenerative disease, and MSCs would be a strong candidate for such a treatment strategy.
So far, a large number of studies have verified the effect of MSCs on multi-target clearance of Aβ amyloid from in vitro and in vivo, but the tumorigenicity of MSCs have also been widely discussed and become one of the most important risks of clinical treatment (Torsvik et al., 2010). At present, there is no report that MSCs induce tumors. However, due to the self-proliferation and differentiation of MSCs are similar to cancer cells, the risk of tumorigenesis cannot be ignored. Therefore, future research will focus on identifying specific small molecules in MSCs that regulate neuroinflammation and induce autophagy to enhance Aβ clearance, which is significant to the development of clinical targets for AD. In addition, although the therapeutic effect of MSCs on AD has been gradually confirmed, to form a systematic treatment system, we must fully consider factors such as the optimal MSCs injection time point, cell concentration, and injection route. We are convinced that, although MSCs research is still in its infancy, the MSCs still have a great deal of hope for patients with AD.
Availability of Data and Materials: Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Authors’ Contribution: The authors confirm contribution to the paper as follows: paper conception and design: Ruxin Zhang, Chunfang Wang; data collection: Ruxin Zhang, Chenggang Li, Ruochen Du; draft manuscript preparation: Ruxin Zhang, Yitong Yuan, Bichun Zhao, Yujuan Zhang. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: This work was supported by a grant from the National Science Foundation of China (No. 82001326), Key R&D project of Shanxi Province (201803D31068) and Applied Basic Research Project of Shanxi Province (201901D211319; 201901D111384); Science and Technology Innovation Project of Colleges and Universities of Shanxi Province (2019L0445; 2019L0418).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
An Y, Liu WJ, Xue P, Ma Y, Zhang LQ, Zhu B, Qi M, Li LY, Zhang YJ, Wang QT (2018). Autophagy promotes MSC-mediated vascularization in cutaneous wound healing via regulation of VEGF secretion. Cell Death & Disease 9: 58. [Google Scholar]
Ariga T, McDonald MP, Yu RK (2008). Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease–A review. Journal of Lipid Research 49: 1157–1175. [Google Scholar]
Behl C (2005). Oxidative stress in Alzheimer’s disease: Implications for prevention and therapy. Sub-Cellular Biochemistry 38: 65–78. [Google Scholar]
Boche D, Perry VH, Nicoll JA (2013). Review: Activation patterns of microglia and their identification in the human brain. Neuropathology and Applied Neurobiology 39: 3–18. [Google Scholar]
Caruso M, Parolini O (2015). Multipotent mesenchymal stromal cell-based therapies: Regeneration versus repair. In: Bhattacharya N, Stubblefield PG (eds.Regenerative Medicine: Using Non-Fetal Sources of Stem Cells, pp. 3–16. London: Springer. [Google Scholar]
Cecchi C, Evangelisti E, Cascella R, Zampagni M, Benvenuti S et al. (2011). Neuronal differentiation of human mesenchymal stromal cells increases their resistance to Aβ42 aggregate toxicity. Journal of Alzheimer’s Disease 27: 651–664. [Google Scholar]
Chen HX, Liang FC, Gu P, Xu BL, Xu HJ, Wang WT, Hou JY, Xie DX, Chai XQ, An SJ (2020a). Exosomes derived from mesenchymal stem cells repair a Parkinson’s disease model by inducing autophagy. Cell Death & Disease 11: 288. [Google Scholar]
Chen Y, Shen J, Ke K, Gu X (2020b). Clinical potential and current progress of mesenchymal stem cells for Parkinson’s disease: A systematic review. Neurological Sciences: Official Journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 41: 1051–1061. [Google Scholar]
Coffey EE, Beckel JM, Laties AM, Mitchell CH (2014). Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 263: 111–124. [Google Scholar]
Cunningham C (2013). Microglia and neurodegeneration: The role of systemic inflammation. Glia 61: 71–90. [Google Scholar]
Czubowicz K, Jęśko H, Wencel P, Lukiw WJ, Strosznajder RP (2019). The role of ceramide and sphingosine-1-phosphate in Alzheimer’s disease and other neurodegenerative disorders. Molecular Neurobiology 56: 5436–5455. [Google Scholar]
de Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL (2007). Aβ oligomers induce neuronal oxidative stress through an n-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. Journal of Biological Chemistry 282: 11590–11601. [Google Scholar]
de la Mont SM, Wands JR (2006). Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. Journal of Alzheimers Disease 9: 167–181. [Google Scholar]
Ding M, Shen Y, Wang P, Xie Z, Xu S et al. (2018). Exosomes isolated from human umbilical cord mesenchymal stem cells alleviate neuroinflammation and reduce amyloid-β deposition by modulating microglial activation in Alzheimer’s disease. Neurochemical Research 43: 2165–2177. [Google Scholar]
Edbauer D, Willem M, Lammich S, Steiner H, Haass C (2002). Insulin-degrading enzyme rapidly removes the β-amyloid precursor protein intracellular domain (AICD). Journal of Biological Chemistry 277: 13389–13393. [Google Scholar]
Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S (2003). Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proceedings of the National Academy of Sciences of the United States of America 100: 4162–4167. [Google Scholar]
Fujikake N, Shin M, Shimizu S (2018). Association between autophagy and neurodegenerative diseases. Frontiers in Neuroscience 12: 255. [Google Scholar]
Godoy MA, Saraiva LM, de Carvalho LRP, Vasconcelos-Dos-Santos A, Beiral HJV et al. (2018). Mesenchymal stem cells and cell-derived extracellular vesicles protect hippocampal neurons from oxidative stress and synapse damage induced by amyloid-β oligomers. Journal of Biological Chemistry 293: 1957–1975. [Google Scholar]
Götz J, Chen F, van Dorpe J, Nitsch RM (2001). Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 293: 1491–1495. [Google Scholar]
Greeve I, Hermans-Borgmeyer I, Brellinger C, Kasper D, Gomez-Isla T et al. (2000). The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. Journal of Neuroscience: The Official Journal of the Society for Neuroscience 20: 7345–7352. [Google Scholar]
Gugliandolo A, Bramanti P, Mazzon E (2019). Mesenchymal stem cells: A potential therapeutic approach for amyotrophic lateral sclerosis? Stem Cells International 2019: 3675627. [Google Scholar]
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889. [Google Scholar]
He X, Huang Y, Li B, Gong CX, Schuchman EH (2010). Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiology of Aging 31: 398–408. [Google Scholar]
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F et al. (2015a). Neuroinflammation in Alzheimer’s disease. Lancet Neurology 14: 388–405. [Google Scholar]
Heneka MT, Golenbock DT, Latz E (2015b). Innate immunity in Alzheimer’s disease. Nature Immunology 16: 229. [Google Scholar]
Hong S, Stevens B (2016). Microglia: Phagocytosing to clear, sculpt, and eliminate. Developmental Cell 38: 126–128. [Google Scholar]
Jaeger PA, Pickford F, Sun CH, Lucin KM, Masliah E, Wyss-Coray T (2010). Regulation of amyloid precursor protein processing by the beclin 1 complex. PLoS One 5: e11102. [Google Scholar]
Jiao H, Shi K, Zhang WJ, Yang L, Yang L, Guan FX, Yang B (2016). Therapeutic potential of human amniotic membrane-derived mesenchymal stem cells in APP transgenic mice. Oncology Letters 12: 1877–1883. [Google Scholar]
Karlawish JH, Casarett DJ, James BD, Xie SX, Kim SY (2005). The ability of persons with Alzheimer disease (AD) to make a decision about taking an AD treatment. Neurology 64: 1514. [Google Scholar]
Kim DH, Lee D, Lim H, Choi SJ, Oh W, Yang YS, Chang JH, Jeon HB (2018a). Effect of growth differentiation factor-15 secreted by human umbilical cord blood-derived mesenchymal stem cells on amyloid β levels in vitro and in vivo models of Alzheimer’s disease. Biochemical and Biophysical Research Communications 504: 933–940. [Google Scholar]
Kim DH, Lim H, Lee D, Choi SJ, Oh W, Yang YS, Oh JS, Hwang HH, Jeon HB (2018b). Thrombospondin-1 secreted by human umbilical cord blood-derived mesenchymal stem cells rescues neurons from synaptic dysfunction in Alzheimer’s disease model. Scientific Reports 8: 354. [Google Scholar]
Kim DY, Choi SH, Lee JS, Kim HJ, Kim HN, Lee JE, Shin JY, Lee PH (2020a). Feasibility and efficacy of intra-arterial administration of embryonic stem cell derived-mesenchymal stem cells in animal model of Alzheimer’s disease. Journal of Alzheimer’s Disease 76: 1281–1296. [Google Scholar]
Kim J, Lee Y, Lee S, Kim K, Song M, Lee J (2020). Mesenchymal stem cell therapy and Alzheimer’s disease: Current status and future perspectives. Journal of Alzheimer’s Disease 77: 1–14. [Google Scholar]
Kim JY, Kim DH, Kim DS, Kim JH, Jeong SY, Jeon HB, Lee EH, Chang JW (2010). Galectin-3 secreted by human umbilical cord blood-derived mesenchymal stem cells reduces amyloid-β42 neurotoxicity in vitro. FEBS Letters 100: 415a. [Google Scholar]
Kim KS, Kim HS, Park JM, Kim HW, Park MK, Lee HS, Lim DS, Lee TH, Chopp M, Moon J (2013). Long-term immunomodulatory effect of amniotic stem cells in an Alzheimer’s disease model. Neurobiology of Aging 34: 2408–2420. [Google Scholar]
Lee H, Lee JK, Min WK, Bae JH, He X, Schuchman EH, Bae JS, Jin HK (2010a). Bone marrow-derived mesenchymal stem cells prevent the loss of Niemann-Pick type C mouse Purkinje neurons by correcting sphingolipid metabolism and increasing sphingosine-1-phosphate. Stem Cells 28: 821–831. [Google Scholar]
Lee HJ, Lee JK, Lee H, Carter JE, Chang JW et al. (2012a). Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiology of Aging 33: 588–602. [Google Scholar]
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS et al. (2010c). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141: 1146–1158. [Google Scholar]
Lee JK, Jin HK, Bae JS (2009). Bone marrow-derived mesenchymal stem cells reduce brain amyloid-β deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neuroscience Letters 450: 136–141. [Google Scholar]
Lee JK, Jin HK, Endo S, Schuchman EH, Carter JE, Bae JS (2010b). Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-β deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells 28: 329–343. [Google Scholar]
Lee JK, Schuchman EH, Jin HK, Bae JS (2012b). Soluble CCL5 derived from bone marrow-derived mesenchymal stem cells and activated by amyloid β ameliorates Alzheimer’s disease in mice by recruiting bone marrow-induced microglia immune responses. Stem Cells 30: 1544–1555. [Google Scholar]
Lee S, Sato Y, Nixon RA (2011). Primary lysosomal dysfunction causes cargo-specific deficits of axonal transport leading to Alzheimer-like neuritic dystrophy. Autophagy 7: 1562–1563. [Google Scholar]
Li F, Calingasan NY, Yu F, Mauck WM, Toidze M et al. (2004). Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. Journal of Neurochemistry 89: 1308–1312. [Google Scholar]
Li T, Xia M, Gao Y, Chen Y, Xu Y (2015). Human umbilical cord mesenchymal stem cells: An overview of their potential in cell-based therapy. Expert Opinion on Biological Therapy 15: 1293–1306. [Google Scholar]
Li W, Li K, Gao J, Yang Z (2018). Autophagy is required for human umbilical cord mesenchymal stem cells to improve spatial working memory in APP/PS1 transgenic mouse model. Stem Cell Research & Therapy 9: 9. [Google Scholar]
Lim H, Lee D, Choi WK, Choi SJ, Oh W et al. (2020). Galectin-3 secreted by human umbilical cord blood-derived mesenchymal stem cells reduces aberrant tau phosphorylation in an Alzheimer disease model. Stem Cells International 2020: 8878412. [Google Scholar]
Losurdo M, Pedrazzoli M, D’Agostino C, Elia CA, Massenzio F et al. (2020). Intranasal delivery of mesenchymal stem cell-derived extracellular vesicles exerts immunomodulatory and neuroprotective effects in a 3xTg model of Alzheimer’s disease. Stem Cells Translational Medicine 9: 1068–1084. [Google Scholar]
Ma B, Cao W, Li W, Gao C, Qi Z et al. (2014). Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Research 24: 912–924. [Google Scholar]
Ma T, Gong K, Ao Q, Yan Y, Song B, Huang H, Zhang X, Gong Y (2013). Intracerebral transplantation of adipose-derived mesenchymal stem cells alternatively activates microglia and ameliorates neuropathological deficits in Alzheimer’s disease mice. Cell Transplantation 22: S113–126. [Google Scholar]
Ma X, Huang M, Zheng M, Dai C, Song Q et al. (2020). ADSCs-derived extracellular vesicles alleviate neuronal damage, promote neurogenesis and rescue memory loss in mice with Alzheimer’s disease. Journal of Controlled Release: Official Journal of the Controlled Release Society 327: 688–702. [Google Scholar]
Maceyka M, Harikumar KB, Milstien S, Spiegel S (2012). Sphingosine-1-phosphate signaling and its role in disease. Trends in Cell Biology 22: 50–60. [Google Scholar]
Mandrekar-Colucci S, Landreth GE (2010). Microglia and inflammation in Alzheimer’s disease. CNS & Neurological Disorders Drug Targets 9: 156–167. [Google Scholar]
Marfia G, Navone SE, Hadi LA, Paroni M, Berno V et al. (2016). The adipose mesenchymal stem cell secretome inhibits inflammatory responses of microglia: Evidence for an involvement of sphingosine-1-phosphate signalling. Stem Cells and Development 25: 1095–1107. [Google Scholar]
Masters CL, Selkoe DJ (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine 2: a006262. [Google Scholar]
Matsuzaki K (2020). Aβ-ganglioside interactions in the pathogenesis of Alzheimer’s disease. Biochimica et Biophysica Acta. Biomembranes 1862: 183233. [Google Scholar]
Mehrabadi S, Motevaseli E, Sadr SS, Moradbeygi K (2020). Hypoxic-conditioned medium from adipose tissue mesenchymal stem cells improved neuroinflammation through alternation of toll like receptor (TLR) 2 and TLR4 expression in model of Alzheimer’s disease rats. Behavioural Brain Research 379: 112362. [Google Scholar]
Mezey E, Key S, Vogelsang G, Szalayova I, Lange GD, Crain B (2003). Transplanted bone marrow generates new neurons in human brains. Proceedings of the National Academy of Sciences of the United States of America 100: 1364–1369. [Google Scholar]
Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL (2003). Amyloid-β peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proceedings of the National Academy of Sciences of the United States of America 100: 6221–6226. [Google Scholar]
Mines M, Ding Y, Fan GH (2007). The many roles of chemokine receptors in neurodegenerative disorders: Emerging new therapeutical strategies. Current Medicinal Chemistry 14: 2456–2470. [Google Scholar]
Misonou H, Morishima-Kawashima M, Ihara Y (2000). Oxidative stress induces intracellular accumulation of amyloid β-protein (Aβ) in human neuroblastoma cells. Biochemistry 39: 6951–6959. [Google Scholar]
Mo SJ, Zhong Q, Zhou YF, Deng DB, Zhang XQ (2012). Bone marrow-derived mesenchymal stem cells prevent the apoptosis of neuron-like PC12 cells via erythropoietin expression. Neuroscience Letters 522: 92–97. [Google Scholar]
Moonga J, Likupe G (2016). A systematic literature review on nurses’ and health care support workers’ experiences of caring for people with dementia on orthopaedic wards. Journal of Clinical Nursing 25: 1789–1804. [Google Scholar]
Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW (2002). A CD36-initiated signaling cascade mediates inflammatory effects of β-amyloid. Journal of Biological Chemistry 277: 47373–47379. [Google Scholar]
Morgan BP (2018). Complement in the pathogenesis of Alzheimer’s disease. Seminars in Immunopathology 40: 113–124. [Google Scholar]
Neniskyte U, Fricker M, Brown GC (2016). Amyloid β induces microglia to phagocytose neurons via activation of protein kinase Cs and NADPH oxidase. International Journal of Biochemistry & Cell Biology 81: 346–355. [Google Scholar]
Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M (2009). APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457: 981–989. [Google Scholar]
Park BN, Kim JH, Lim TS, Park SH, Kim TG, Yoon BS, Son KS, Yoon JK, An YS (2020a). Therapeutic effect of mesenchymal stem cells in an animal model of Alzheimer’s disease evaluated by β-amyloid positron emission tomography imaging. Australian and New Zealand Journal of Psychiatry 54: 000486742091746. [Google Scholar]
Park H, Kang JH, Lee S (2020b). Autophagy in neurodegenerative diseases: A hunter for aggregates. International Journal of Molecular Sciences 21: 3369. [Google Scholar]
Parr AM, Tator CH, Keating A (2007). Bone marrow-derived mesenchymal stromal cells for the repair of central nervous system injury. Bone Marrow Transplant 40: 609–619. [Google Scholar]
Piccinini M, Scandroglio F, Prioni S, Buccinnà B, Loberto N et al. (2010). Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Molecular Neurobiology 41: 314–340. [Google Scholar]
Prockop DJ, Gregory CA, Spees JL (2003). One strategy for cell and gene therapy: Harnessing the power of adult stem cells to repair tissues. Proceedings of the National Academy of Sciences of the United States of America 100: 11917–11923. [Google Scholar]
Quiroz-Baez R, Rojas E, Arias C (2009). Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of α-, β- and γ-secretase expression. Neurochemistry International 55: 662–670. [Google Scholar]
Ransohoff RM, Liu L, Cardona AE (2007). Chemokines and chemokine receptors: Multipurpose players in neuroinflammation. International Review of Neurobiology 82: 187–204. [Google Scholar]
Reyhani S, Abbaspanah B, Mousavi SH (2020). Umbilical cord-derived mesenchymal stem cells in neurodegenerative disorders: From literature to clinical practice. Regenerative Medicine 15: 1561–1578. [Google Scholar]
Rocchi A, Yamamoto S, Ting T, Fan Y, Sadleir K et al. (2017). A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genetics 13: e1006962. [Google Scholar]
Rogers J, Strohmeyer R, Kovelowski CJ, Li R (2002). Microglia and inflammatory mechanisms in the clearance of amyloid β peptide. Glia 40: 260–269. [Google Scholar]
Sadan O, Shemesh N, Barzilay R, Dadon-Nahum M, Blumenfeld-Katzir T, Assaf Y, Yeshurun M, Djaldetti R, Cohen Y, Melamed E (2012). Mesenchymal stem cells induced to secrete neurotrophic factors attenuate quinolinic acid toxicity: A potential therapy for Huntington’s disease. Experimental Neurology 234: 417–427. [Google Scholar]
Saftig P, Hartmann D, Annaert W, Craessaerts K, Van LF, De SB (1998). The processing of the Amyloid-Precursor-Protein in presenilin-1 deficient neurons. Molecular Biology of Alzheimer’s Disease 1: 235–246. [Google Scholar]
Salem AM, Ahmed HH, Atta HM, Ghazy MA, Aglan HA (2015). Potential of bone marrow mesenchymal stem cells in management of Alzheimer’s disease in female rats. Cell Biology International 38: 1367–1383. [Google Scholar]
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W et al. (2014). Bapineuzumab 301 and 302 Clinical Trial Investigators, Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. New England Journal of Medicine 370: 322–333. [Google Scholar]
Shin JY, Park HJ, Kim HN, Oh SH, Bae JS, Ha H, Lee PH (2014). Mesenchymal stem cells enhance autophagy and increase β-amyloid clearance in Alzheimer disease models. Autophagy 10: 32–44. [Google Scholar]
Si YL, Zhao YL, Hao HJ, Fu XB, Han WD (2011). MSCs: Biological characteristics, clinical applications and their outstanding concerns. Ageing Research Reviews 10: 93–103. [Google Scholar]
Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G (2000). Oxidative stress in Alzheimer’s disease. BBA—Molecular Basis of Disease 1502: 139–144. [Google Scholar]
Suresh SN, Chakravorty A, Giridharan M, Garimella L, Manjithaya R (2020). Pharmacological tools to modulate autophagy in neurodegenerative diseases. Journal of Molecular Biology 432: 2822–2842. [Google Scholar]
Swaminathan G, Zhu W, Plowey ED (2016). BECN1/Beclin 1 sorts cell-surface APP/amyloid β precursor protein for lysosomal degradation. Autophagy 12: 2404–2419. [Google Scholar]
Takasugi N, Sasaki T, Suzuki K, Osawa S, Isshiki H et al. (2011). BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. Journal of Neuroscience 7: S411. [Google Scholar]
Torsvik A, Røsland GV, Svendsen A, Molven A, Immervoll H et al. (2010). Spontaneous malignant transformation of human mesenchymal stem cells reflects cross-contamination: Putting the research field on track-letter. Cancer Research 70: 6393–6396. [Google Scholar]
Wang SQ, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS (2010). Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. Journal of Neurochemistry 115: 47–57. [Google Scholar]
Wang SS, Jia J, Wang Z (2018). Mesenchymal stem cell-derived extracellular vesicles suppresses iNOS expression and ameliorates neural impairment in Alzheimer’s disease mice. Journal of Alzheimer’s Disease 61: 1005–1013. [Google Scholar]
Whyte LS, Lau AA, Hemsley KM, Hopwood JJ, Sargeant TJ (2017). Endo-lysosomal and autophagic dysfunction: A driving factor in Alzheimer’s disease? Journal of Neurochemistry 140: 703–717. [Google Scholar]
Xiao HL, Jackson S, Seaman M, Brown K, Levine B (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672. [Google Scholar]
Xu Z, Nan W, Zhang X, Sun Y, Yang J et al. (2018). Umbilical cord mesenchymal stem cells conditioned medium promotes Aβ25-35 phagocytosis by modulating autophagy and Aβ-degrading enzymes in BV2 cells. Journal of Molecular Neuroscience 65: 222–233. [Google Scholar]
Yokokawa K, Iwahara N, Hisahara S, Emoto MC, Saito T et al. (2019). Transplantation of mesenchymal stem cells improves amyloid-β pathology by modifying microglial function and suppressing oxidative stress. Journal of Alzheimer’s Disease 72: 867–884. [Google Scholar]
Zhao Y, Chen X, Wu Y, Wang Y, Li Y, Xiang C (2018). Transplantation of human menstrual blood-derived mesenchymal stem cells alleviates Alzheimer’s disease-like pathology in APP/PS1 transgenic mice. Frontiers in Molecular Neuroscience 11: 140. [Google Scholar]
Zheng XY, Wan QQ, Zheng CY, Zhou HL, Dong XY et al. (2017). Amniotic mesenchymal stem cells decrease Aβ deposition and improve memory in APP/PS1 transgenic mice. Neurochemical Research 42: 2191–2207. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |