DOI:10.32604/biocell.2022.016984
| BIOCELL DOI:10.32604/biocell.2022.016984 | |
| Article |
The chloroplast genome comparative characteristic of artificial breeding tree, a case about Broussonetia kazinoki × Broussonetia papyrifera
1Hunan Research Center of Engineering Technology for Utilization of Environmental and Resources Plant, Central South University of Forestry and Technology, Changsha, 410004, China
2Hunan Urban and Rural Ecological Planning and Restoration Engineering Research Center, Hunan City University, Yiyang, 413000, China
3Key Laboratory of National Forestry and Grassland Administration on Management of Western Forest Bio-Disaster, College of Forestry, Northwest A&F University, Yangling, 712100, China
*Address correspondence to: Zhenggang Xu, xuzhenggang@nwafu.edu.cn
#These authors contributed equally to this work
Received: 16 April 2021; Accepted: 07 June 2021
Abstract: Broussonetia kazinoki × Broussonetia papyrifera (ZJGS) is a hybrid species in Moraceae family, which has a very complicated hybrid origin. The excellent characteristics of fast growth, strong soil and water conservation ability, high leaf protein content and stem fiber content in ZJGS make it both ecological benefits in the mining area and economically valuable. This study aims to further understand ZJGS and other Moraceae taxa through the ZJGS chloroplast (cp) genome structure and the comparison with 12 closely related Moraceae species. Among the 13 Moraceae species, the cp genome length of seven Broussonetia species (ranges from 160,239 bp to 162,594 bp) is larger than that of six Morus species (ranges from 158,459 bp to 159,265 bp). Among the 77 shared protein-coding genes (PCGs) in Moraceae species, the obvious positive selection of Ka/Ks ratios acted on petD and rpl16 genes of B. kazinoki and B. papyrifera, respectively. Phylogenetic analysis based on shared PCGs from 28 species shows that ZJGS is closely related to maternal B. kazinoki. These findings provide data support for the origin of ZJGS hybridization and provide genomic resources for future ZJGS resource development and molecular breeding.
Keywords: Broussonetia kazinoki; Broussonetia papyrifera; Moraceae; Phylogenetic relationship; Maternal inheritance
Abbreviations
| B. papyrifera: | Broussonetia papyrifera |
| bp: | Base pairs |
| cp: | Chloroplast |
| CNR1: | Cell Number Regulator 1 gene |
| DOGMA: | Dual Organellar GenoMe Annotator |
| F. carica: | Ficus carica |
| IRs: | inverted repeats |
| Ka: | the number of nonsynonymous substitutions per non-synonymous site |
| Ks: | the number of synonymous substitutions per synonymous site |
| LSC: | large single copy |
| ML: | maximum likelihood method |
| MP: | maximum parsimony method |
| M. atropurpurea: | Morus atropurpurea |
| NJ: | neighbor-joining method |
| NAD(P)H: | Nicotinamide adenine dinucleotide (reduced) |
| NDH: | NAD(P)H dehydrogenase |
| PCGs: | proteincoding genes |
| SSC: | small single copy |
| rRNA: | Ribosomal RNA |
| SSR: | Simple sequence repeats |
| SPR: | Subtree-Pruning-Regrafting |
| tRNA: | Transfer Ribonucleic Acid |
| ZJGS: | Broussonetia kazinoki × Broussonetia papyrifera |
Ther0e are many hybridization phenomena in nature, which are regarded as effective methods to produce “positive species” (Gowda et al., 2010; Pucher et al., 2016; Tompkins et al., 2006). Hybrid breeding is widely used in forestry production, especially in horticulture. At present, there are already many hybrid breeds used in agriculture and forestry production based on heterosis. The hybrid aspen (Populus tremula × Populus tremuloides) has a higher yield than the parent species and can be quickly regenerated from the root sucker (Liesebach et al., 1999; Rytter, 2002; Rytter and Stener, 2005). Hybrids of Abies genus have the advantage of strong anti-pollution ability and resistance to pests and diseases (Kobliha and Stejskal, 2009; Kobliha et al., 2013). The heterosis is predicted by both positive gene contribution and environment. Hybridization is also an important way to produce gene diversity. Biologists have always focused on the role of hybridization in evolution. In-depth analyses of heterozygous genotypes showed that heterosis mainly came from the accumulation of a large number of rare superior alleles with positive dominance (Huang et al., 2015). Recently, five candidate genes involved in high yield were identified in super hybrid rice LYP9, among which heterozygous segments containing qSS7 and qHD8 showed superiority and contributed to heterosis (Lin et al., 2020). In maize, the silent expression of Cell Number Regulator 1 gene (CNR1) can increase the size of plant and organ and become a direct contributor to heterosis (Guo et al., 2010). The nucleotide sequence of the plastid gene rbcL and the nuclear gene PgiC were used to study the reticulated evolution of the Dryopteris varia complex in Japan and a haplotype not belonging to any existing species was found. It was speculated that it came from an extinct species. This indicates that hybrids may have higher fitness due to heterosis and become an effective way to preserve their genetic diversity (Hori et al., 2014). However, the above genetic information related to hybridization is mostly concentrated in nuclear genome. Studies have shown that there is gene exchange between the chloroplast (cp) genome and the nuclear genome (Jansen et al., 2007; Lin et al., 2017; Sugiura, 2015; Timmis et al., 2004), so the heterosis of nuclear genes may affect the cp genome. Typically, the cp genome is minimal when compared with the plant mitochondrial and nuclear genomes, which has significant advantages in the study of phylogenetic evolution of species.
Cp genome, as an independent genetic unit, is mostly unisexual, although multiple inheritance patterns had been found in the cpDNA of Actinidia (Li et al., 2013). The selection pressure during the evolution of the cp genome is small, which can directly reflect the genetic variation accumulated in the long-term evolution of plants, and can be used to trace the origin and migration of species as well as study genetic diversity (Nadachowska-Brzyska et al., 2015; Timmis et al., 2004; Won, 2019). The cp genome contains a large number of functional genes, which can be divided into three categories: genes related to photosynthesis (Photosystem I: psa; Subunits of Photosystem I: ycf3 and ycf4; Photosystem II: psb), genes related to gene expression (ribosomal RNA genes and transfer RNA genes, etc.), and other genes related to biosynthesis (ATP synthase gene, NADH dehydrogenase gene, etc.). As the two largest genes, ycf1 and ycf2 located at the IR/SC junction and the IRs regions, respectively, which proved to be useful for analyzing cp genome variation in higher plants (Zhang et al., 2013). As the development of sequencing technology, cp genome sequences are more and more available. The phylogenetic analyses based on the cp genome are achievable and revealed qualitative and valuable information to confirm the genetic relationship of different species (Moore et al., 2010; Nguyen et al., 2015b). The complete cp genome sequence of Morus cathayana and Morus multicaulis were obtained and compared with other genus Morus, the results indicated that both natural selection and mutational bias have contributed to the codon bias (Kong and Yang, 2017).
Paper mulberry is a member of genus Broussonetia in Moraceae family. The tree displayed the features on fast-growing, easy breeding, resistant to pruning and widely distributing (Morgan and Overholt, 2016; Peng et al., 2015; Yan et al., 2011). Vegetation survey in tailing’s area showed that the tree is an excellent native plant having a comprehensive absorption effect for various heavy metals (Zhao et al., 2014). Meanwhile, the leaves are an important source of pig feeds and some chemical substances are also important resources for medicine (Ryu et al., 2012). Most importantly, as the name implies, because of its long fiber and ease of preparation, paper mulberry contributed to the invention of papermaking and then was also used in barkcloth (Moncada et al., 2013). Based on the multiple uses of paper mulberry, paper mulberry has historically been an important economic tree species and even accompanied by human expansion (Chang et al., 2015). Now, there is some confusion in the name of paper mulberry. Paper mulberry, Broussonetia papyrifera, B. kazinoki × B. papyrifera are all used to called the tree, while it may not the same tree (Xu et al., 2018). In recent years, a hybrid variety for paper mulberry was bred (Peng et al., 2014; Won, 2019). The breeding process is showed in Fig. 1 and in order to standardize the name, we called it as the hybrid paper mulberry (ZJGS) in the study. ZJGS is a hybrid between B. kazinoki and B. papyrifera, whose maternal lineage is B. kazinoki and paternal is B. papyrifera, respectively (Ni et al., 2020; Peng et al., 2014; Won, 2019). With a long breeding process of more than ten years, the researchers first used modern breeding technology and then loaded the hybrid materials into the spacecraft and bred by space mutation. The mutant materials were optimized and cultivated after the spacecraft returned to the ground, and finally hybrid plants with excellent traits were obtained. However, B. kazinoki was found to be probably a naturally occurring hybrid, with B. monoica as the female parent and B. papyrifera as the male parent (Won, 2019). The complicated hybridization process has blurred the genetic background of paper mulberry and hindered the determination of the classification status of the tree and its utilization.
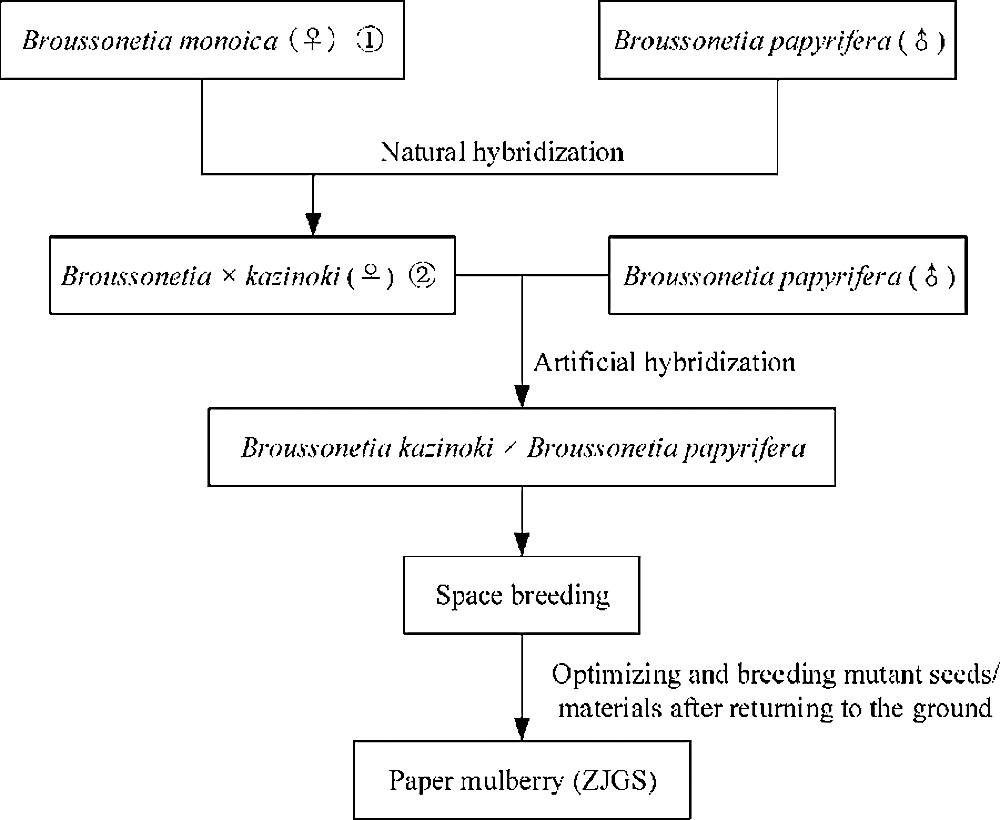
Figure 1: Schematic diagram of ZJGS breeding. Note: The Broussonetia monoica ① was named as Broussonetia kaempferi in the flora of China. The Broussonetia kazinoki ② was also called as B. × hanjiana in Korea and B. × kazinoki in Japan.
Due to the tree nuclear genome is always large, a few forest tree genomes have been sequenced. Among the genetic resources of ZJGS, only nuclear genome of B. papyrifera was assembled (Peng et al., 2019). Although we have reported the cp genome structure of ZJGS, comparative analysis of related species is lacking (Xu et al., 2018). The vacancy of molecular genetic information about ZJGS may prevent optimal breeding of the tree and the exploration of adaptability mechanisms. In order to further determine the characteristics of the cp genome during the hybridization process, the cp genome of ZJGS was compared with 12 Moraceae species, including B. papyrifera and B. kazinoki. The research is not only helpful for us to further determine the status of hybrid paper mulberry, but also provide reference value for future molecular markers and breeding of new varieties.
Comparative analyses of cp genome structure
Our research group has reported the complete cp genome of ZJGS, and the sequence was submitted to GeneBank with the accession number of MF496038. The more experiment details can be got from the announcement (Xu et al., 2018). At the same time, 12 Moraceae plants, including six Broussonetia species and six Morus species were selected to compare cp genomes structure with ZJGS. The cp genomes information of 13 Moraceae plants were downloaded from NCBI database and the GenBank accession numbers were listed in Suppl. Table S1. Firstly, the characteristics of cp genomes, such as GC content, length of IRs regions and number of genes were compared manually. Secondly, mVista in Shuffle-LAGAN mode was employed to compare cp genomes of the above selected 13 plants and ZJGS was set as the reference (Brudno et al., 2003). To explore the evolutionary event of ZJGS, the details of IRs junction regions were detected and displayed, for the IRs regions were considered as the most conserved regions and these junctions were regarded as an index of cp genome evolution.
Simple sequence repeats (SSRs) and long repeats sequences analysis
SSRs, which is one of the most widely used molecular marker systems in plant genetic breeding and revealed more polymorphism (Powell et al., 1996; Suo et al., 2016; Umadevi et al., 2014). The SSRs within the cp genome of each species were predicted by MISA with the parameters were set as: ≥10 for mononucleotides, ≥4 for di-nucleotides, ≥3 for tetra-nucleotides, penta-nucleotides and hexa-nucleotides. The number and details of SSRs were showed. Long repeat sequences, including forward match, reverse match, palindromic match and complementary match were identified by REPuter (Kurtz et al., 2001). The related settings were showed as follows: (1) 90% or greater sequence identity; (2) a minimal repeat size of 20 bp. The graphs were all plotted using software SigmaPlot 12.5.
The Ka/Ks ratio is a good indicator of selective pressure at the sequence level, which can calculate selective pressure within protein coding regions. Here, the Ka/Ks ratio of 77 shared protein-coding genes (PCGs) of 12 Moraceae plants cp genomes were compared with ZJGS. Firstly, the multiple nucleotide sequences of the homologous genes that code for proteins were aligned by MEGA 7 (Kumar et al., 2016), and then the Ka/Ks ratio was calculated using DnaSP v5 (Librado and Rozas, 2009).
In order to clarify the phylogenetic relationship of ZJGS, 27 related species were downloaded from the NCBI for phylogenetic analysis (the GenBank accession numbers were listed in Suppl. Table S1). The multiple sequences were aligned use MAFFT v7 (Katoh and Standley, 2013) in PhyloSuite v1.2.2 (Zhang et al., 2020) with the default parameters strategy and normal alignment mode. ModelFinder (Kalyaanamoorthy et al., 2017) was used to select the best-fit model (GTR + F + I + G4) using BIC criterion. Maximum likelihood (ML) phylogenies were inferred using IQ-TREE (Nguyen et al., 2015a) under the model automatically selected for 100 standard bootstraps, as well as the Shimodaira-Hasegawa-like approximate likelihood-ratio test. Bayesian Inference (BI) phylogenies were inferred using MrBayes 3.2.6 (Ronquist et al., 2012) under GTR + I + G + F model (2 parallel runs, 20,000 generations), in which the initial 25% of sampled data were discarded as burn-in. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) were shown next to the branches.
Comparative analyses of the cp genomes of Moraceae species
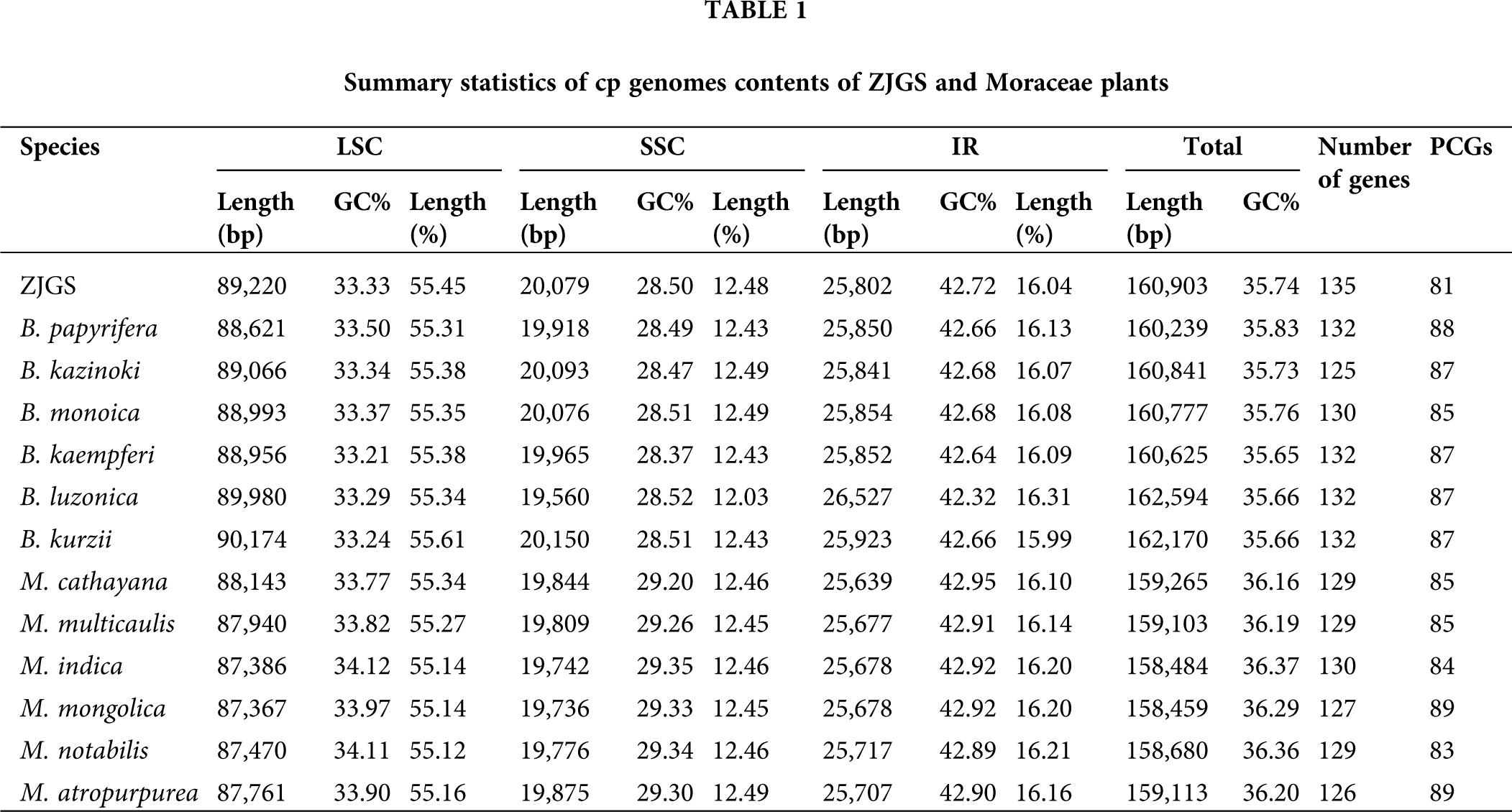
In the Moraceae species of comparison, the cp genome length of all seven Broussonetia (ranges from 160,239 bp to 162,594 bp) is higher than that of the six Morus (ranges from 158,459 bp to 159,265 bp), but the total GC content is relatively lower in the genus Broussonetia, the average value is 35.72%, which in Morus is 36.26% (Table 1). From the perspective of cp genome length, the species that are closer to ZJGS are B. papyrifera, B. kazinoki, B. monoica and B. kaempferi, and the GC content of B. kazinoki is most similar to ZJGS. The length ratio of the IRs regions in ZJGS cp genome is the lowest among all the comparative species, which is 16.04%. In addition, the ZJGS cp genome contains the largest number of genes, while the number of genes that direct synthetic proteins is the least, only 81.
CpDNA variation and conservation of ZJGS
In order to detect the global sequence variability of the Moraceae cp genome, the sequence of ZJGS was used as a reference to compare with the other Moraceae species (Fig. 2). It is found that the ZJGS cp genomic sequence has the highest similarity with B. kazinoki and B. monoica by comparison. Furthermore, the sequence of its paternal B. papyrifera exhibits higher divergence. These highly different regions include matK, rps16, atpF, rpoC2, rpoC1, ycf3, clpP, ndhF, ccsA, ndhA and ycf1. It has become apparent to us that these genes are good sources for interspecies phylogenetic analysis and evolutionary studies. There are also some genes that are relatively conserved among these species, such as atpH, petN, psbE, rpl2, psbH, psbM and rps7. As a whole, the non-coding sequence of the Moraceae plants is more divergent than the coding sequence, and the sequence variation in the IRs regions is smaller than that in the LSC and SSC regions, which also shows that the IRs regions are more conservative than the LSC and SSC regions.
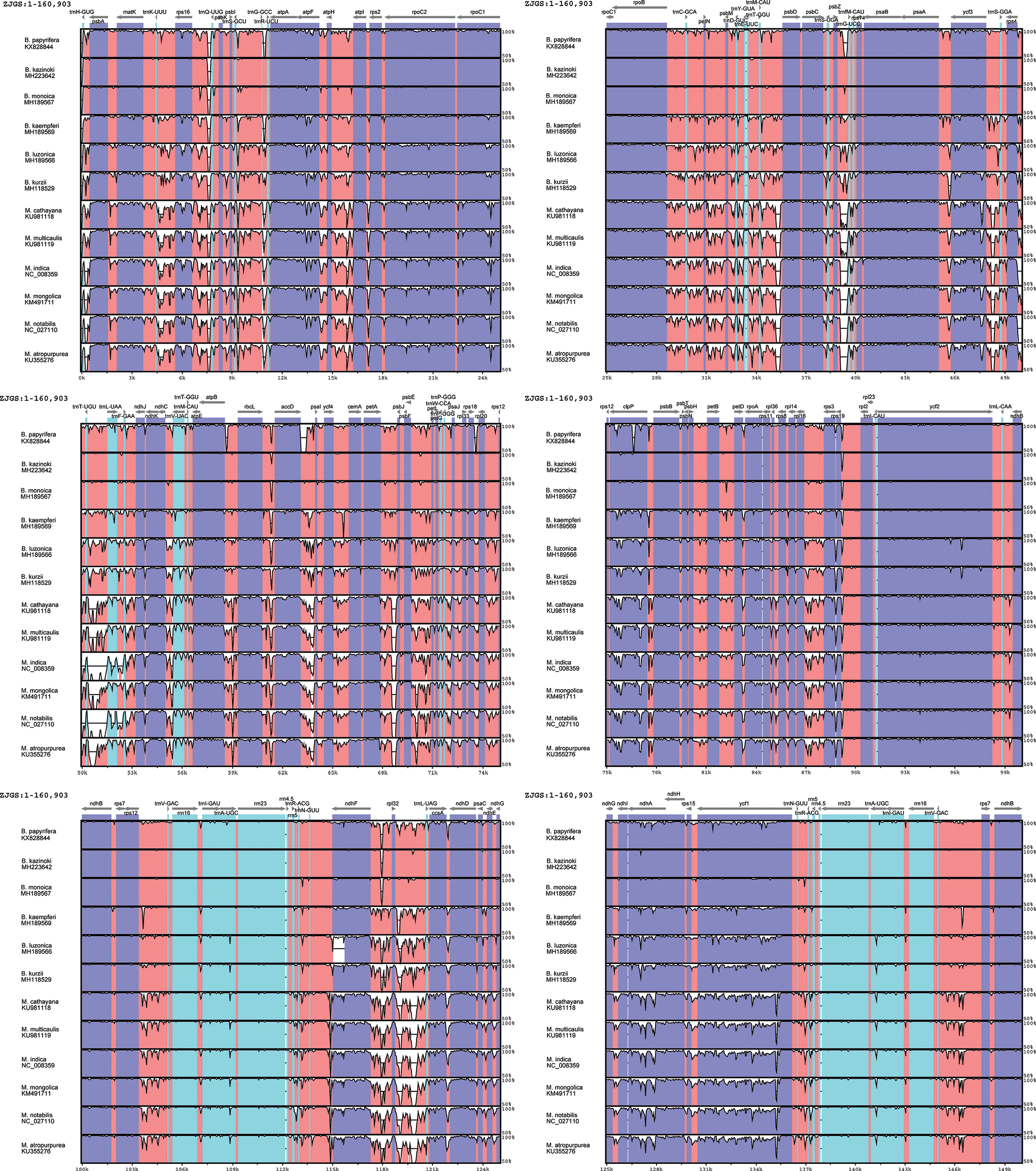
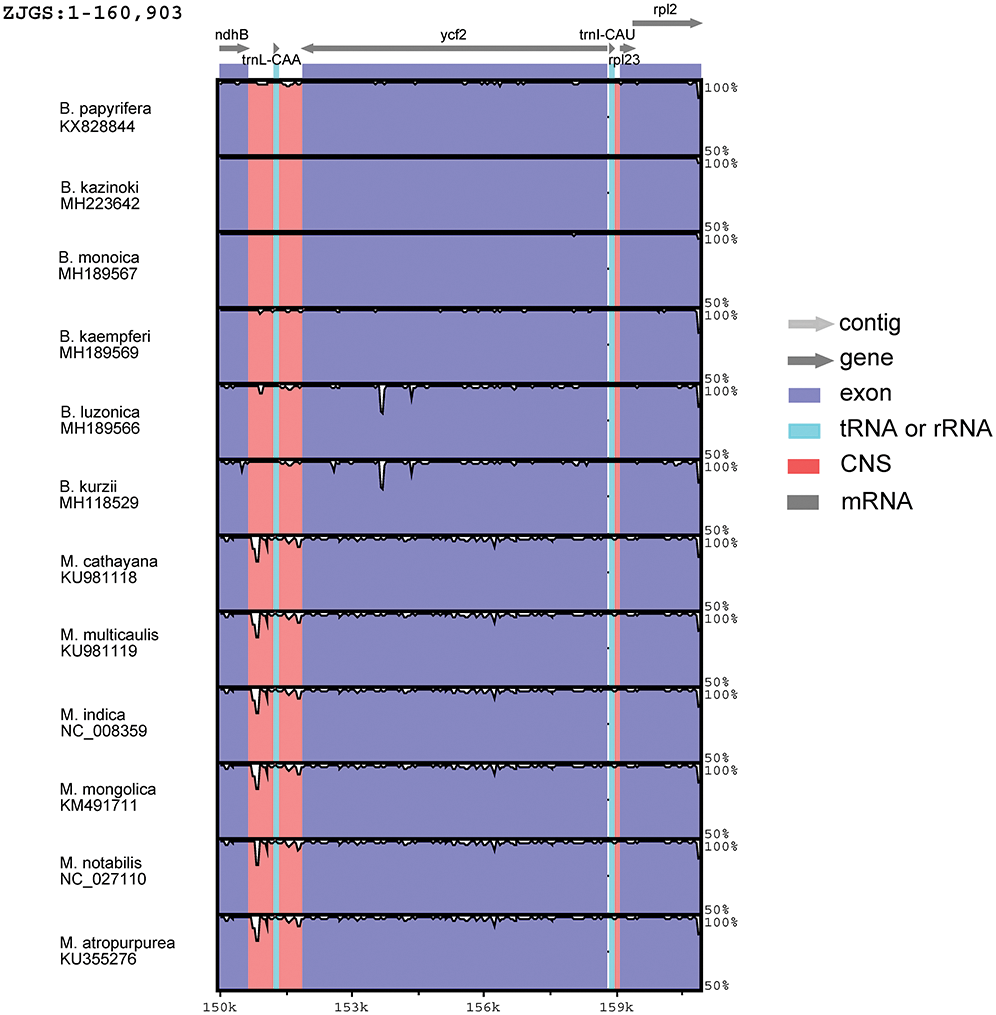
Figure 2: Comparison of sequence similarity among 13Moraceae plants cp genomes using ZJGS as reference. Arrows indicate the direction of the gene, where exons are shown in blue, tRNA or rRNA genes are shown in green, and CNS represent non-coding sequences, indicated in red.
Intuitive results show that the gene composition in IR/SC junction of ZJGS cp genome is somewhat different from that of several other species (Fig. 3). Among them, the most obvious is that in IRa/SSC region, ycf1 is replaced by trnN in ZJGS cp genome, which is the same as B. monoica, B. kaempferi, B. luzonica and B. kurzii. However, the genetic constitution in the two parent species of ZJGS at the IR/SC junction is similar. In terms of gene length, the rpl2 gene (401 bp) in the ZJGS cp genome is significantly shortened in the IRa region and is more than three times shorter than other species (1,509 bp), which is related to the IRa boundary contraction.
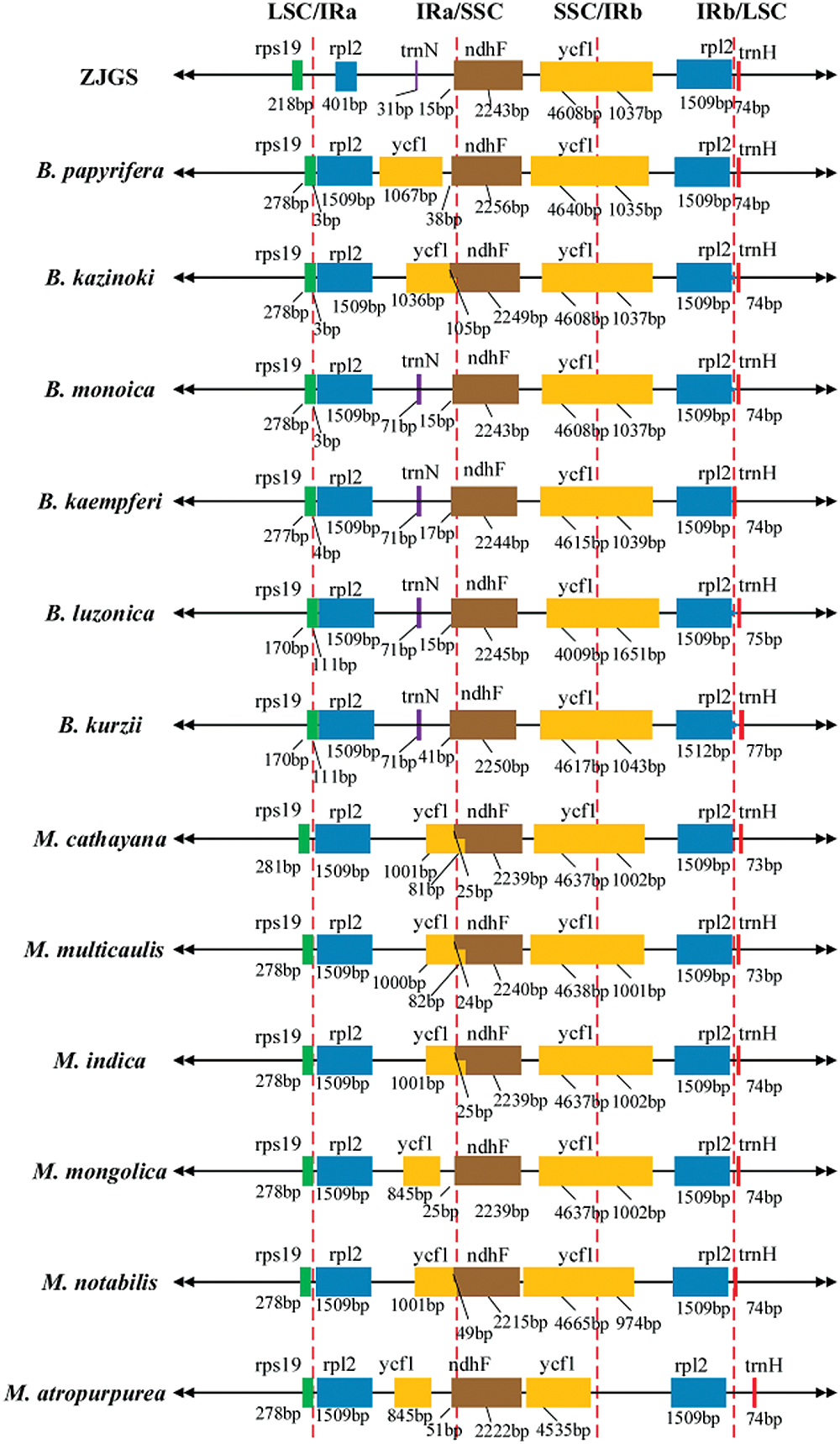
Figure 3: Comparison of ZJGS cp genome with the IR/SC borders of other 12 Moraceae species.
In this study, the SSRs of ZJGS and 12 Moraceae species cp genomes were detected (Fig. 4). Similarly, all cp genomes of Moraceae plants have mononucleotides, dinucleotides, trinucleotides, tetranucleotides and pentanucleotides. While, hexanucleotide repeats are only detected in B. papyrifera and B. kurzii. Moreover, the number of SSRs in Broussonetia species is higher than that of Morus species, except for B. papyrifera. On the whole, ZJGS and its maternal species B. kazinoki have the most similar performance among different types of SSRs (Table 2). Mainly reflects in the following SSRs: C/G, AAT/ATT, AAG/CTT, AAAT/ATTT, AGAT/ATCT, ATCG/ATCG, AAATT/AATTT, AATAG/ATTCT, AATAT/ATATT, and AAATGT/ACATTT, which shows the characteristics of maternal inheritance of ZJGS cpDNA. In order to obtain more genetic regulation information, the long repeat sequences of 13 Moraceae plants were further analyzed (Fig. 5A). Four types of repeats were detected in the cp genome of M. atropurpurea and Broussonetia species except for B. luzonica. And the number of palindromic repeats is the largest, then the type of forward repeat and reverse repeat, the least is the complementary repeat. The forward repeats, palindromic repeats, and complementary repeats in the ZJGS cp genome all contain the most repeating elements. Repetitive sequences of 20–30 bp in length reach 50% or more (Figs. 5B–5E). It can be clearly seen that the long repeat sequences of the ZJGS cp genome is dominant in this interval, and these repeat elements can be used as important genetic resources for the genetics and phylogenetic of the ZJGS population.
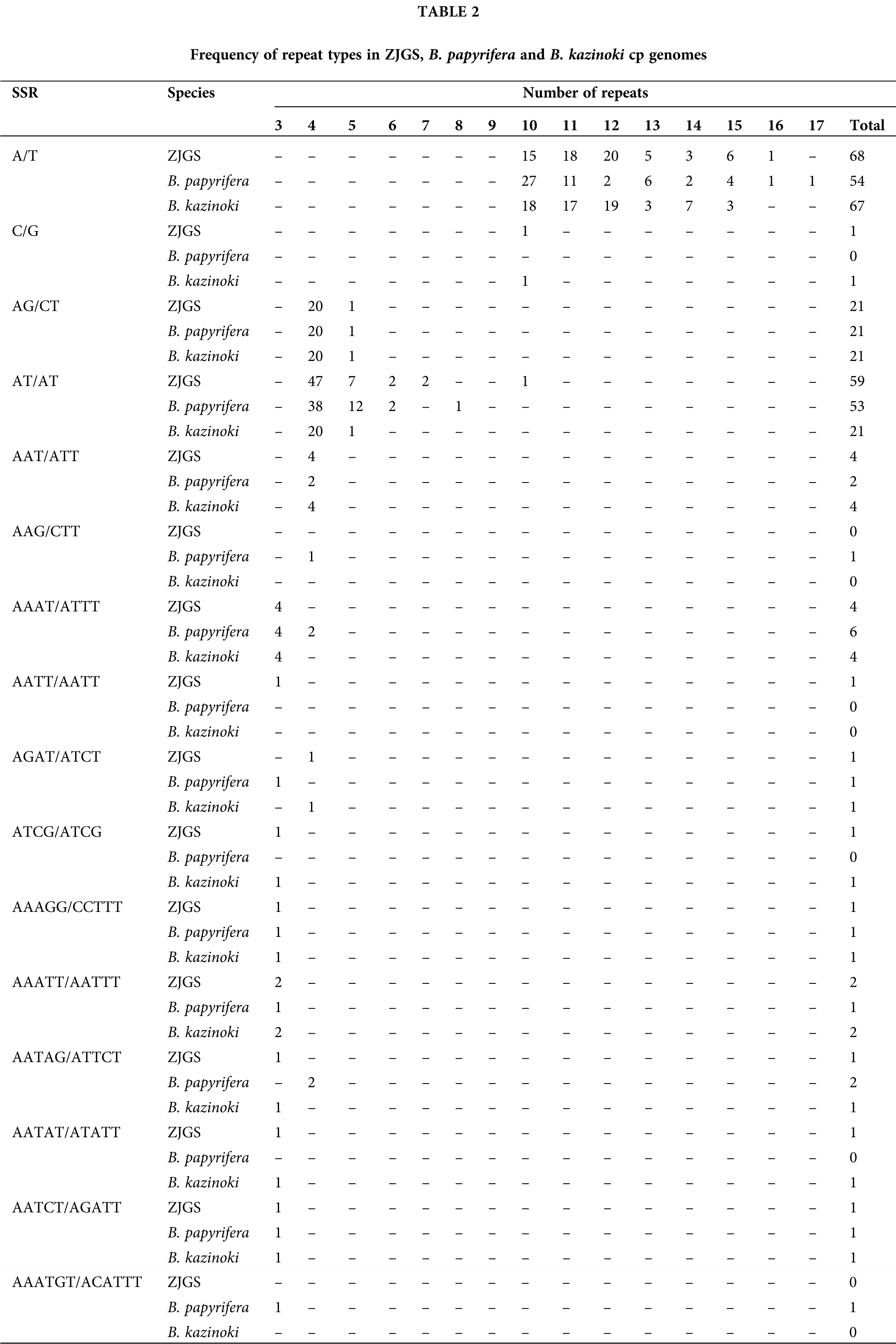
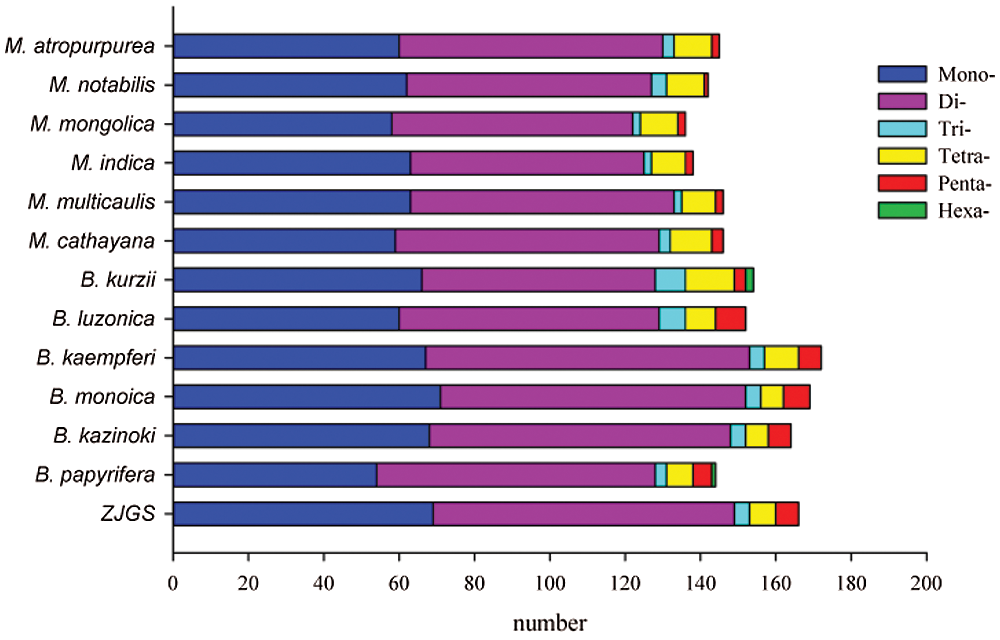
Figure 4: Simple sequence repeats (SSRs) in cp genomes of ZJGS and 12 Moraceae plants.
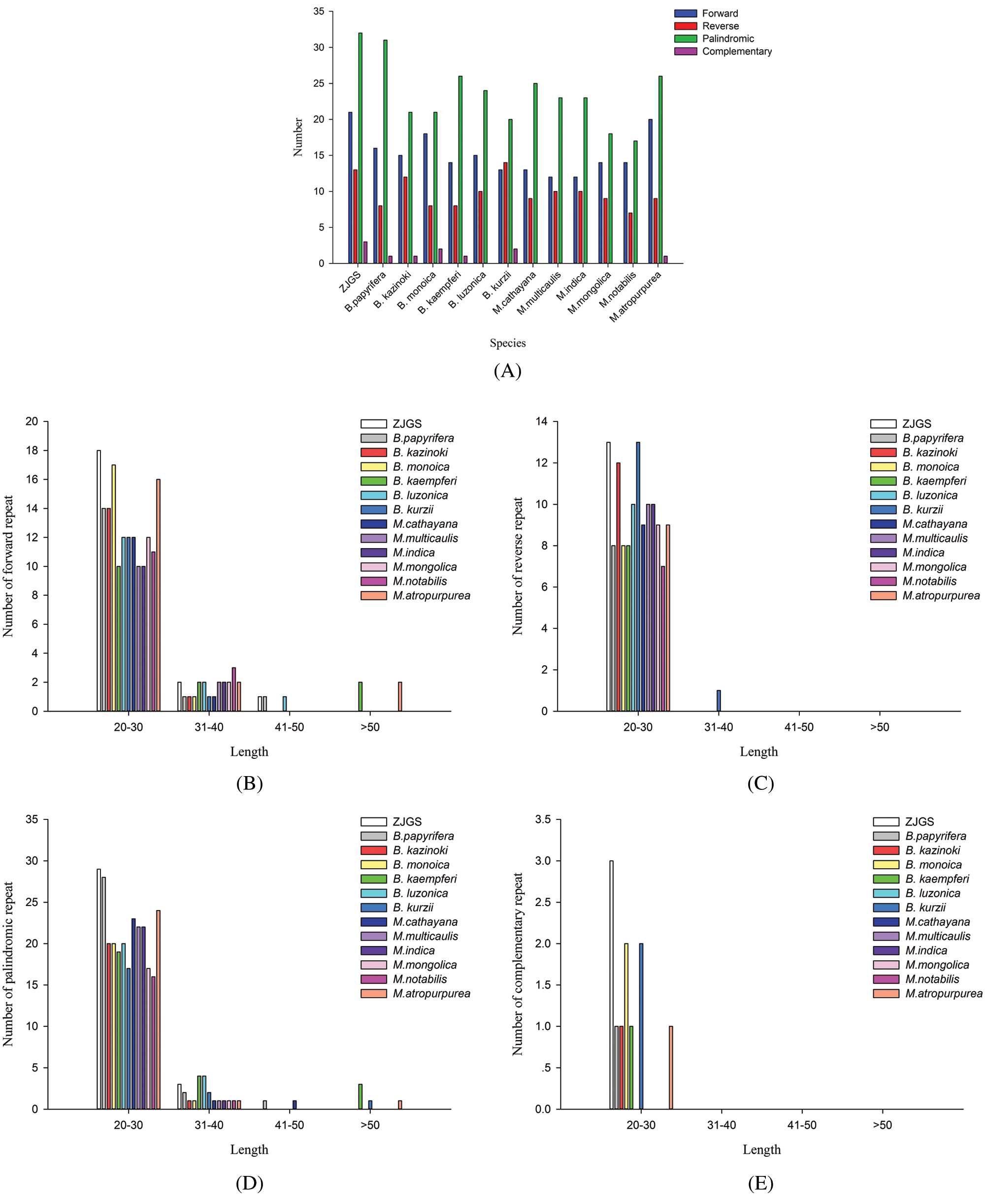
Figure 5: Long repeat sequences in the cp genomes of ZJGS and 12 Moraceae species. (A) The four repetitive sequences. (B) The number of the forward repeat by length. (C) The number of the reverse repeat by length. (D) The number of the palindromic repeat by length. (E) The number of the complementary repeat by length.
The form of sequence evolution: Ka/Ks ratio
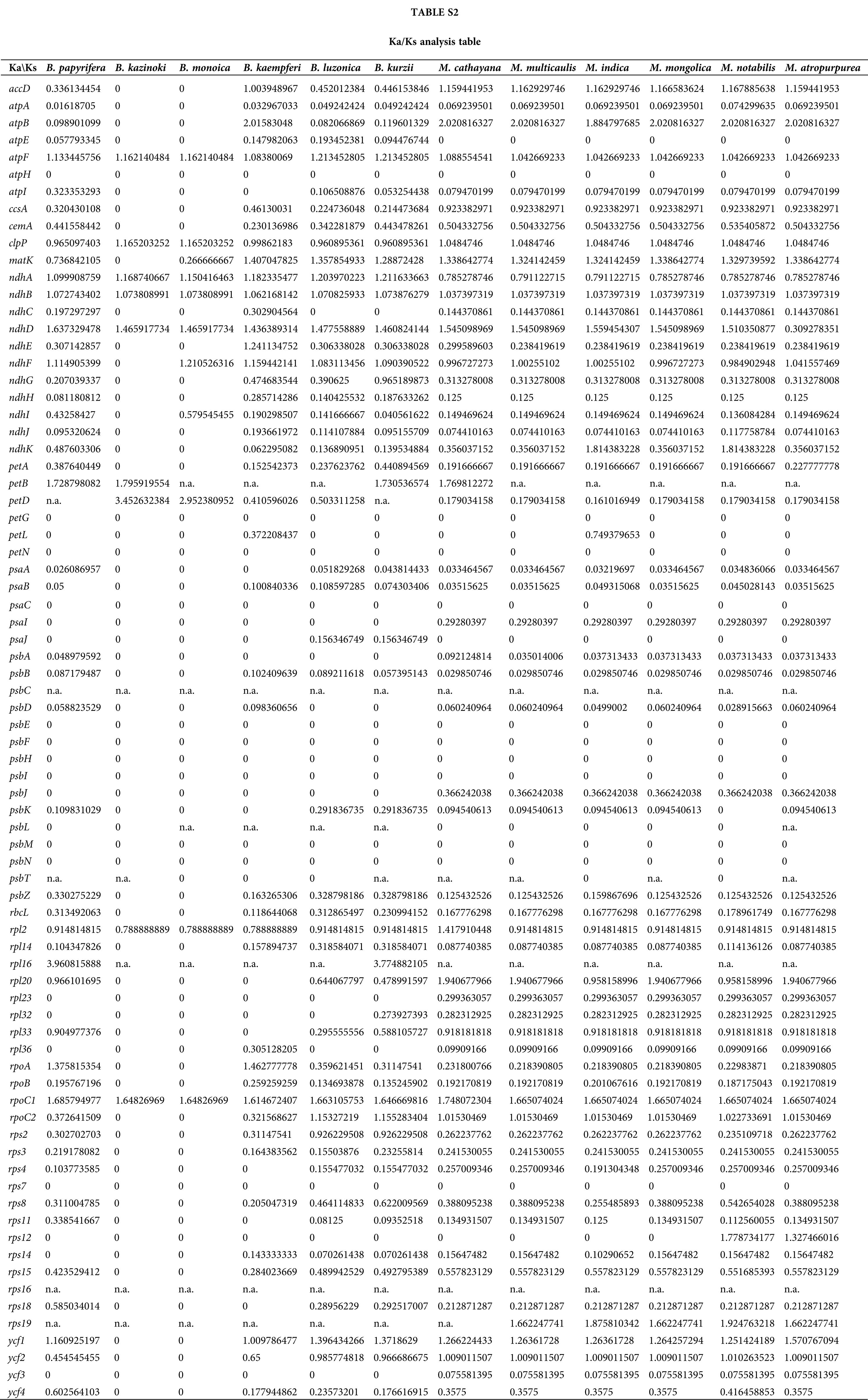
In this study, the cp genome sequence of ZJGS was used as reference, the Ka/Ks ratios of Moraceae species was presented (Fig. 6, Suppl. Table S2). The Ka/Ks ratios of such genes: atpH, petG, petN, psaC, psbE-psbI, psbL-psbT and rps7 is 0, which reflects the strong purification selection and also indicates that these genes are highly conserved. On the contrary, most of the NADH genes (ndhA-ndhK) have higher Ka/Ks ratio, and in the comparison of all species, the Ka/Ks ratio of atpF, ndhB and rpoC1 is >1, these three genes are strongly positive selection. Except for the above three genes, clpP, ndhA, ndhD, petB and petD genes are also positive selection compared with maternal B. kazinoki of ZJGS. While compared with paternal B. papyrifera, ndhA, ndhD, ndhF, petB, rpl16, rpoA and ycf1 genes were positively selected. Among them, petD and rpl16 have the largest Ka/Ks ratio, and both are >3. These two genes are highly evolved and can be used for subsequent gene identification.
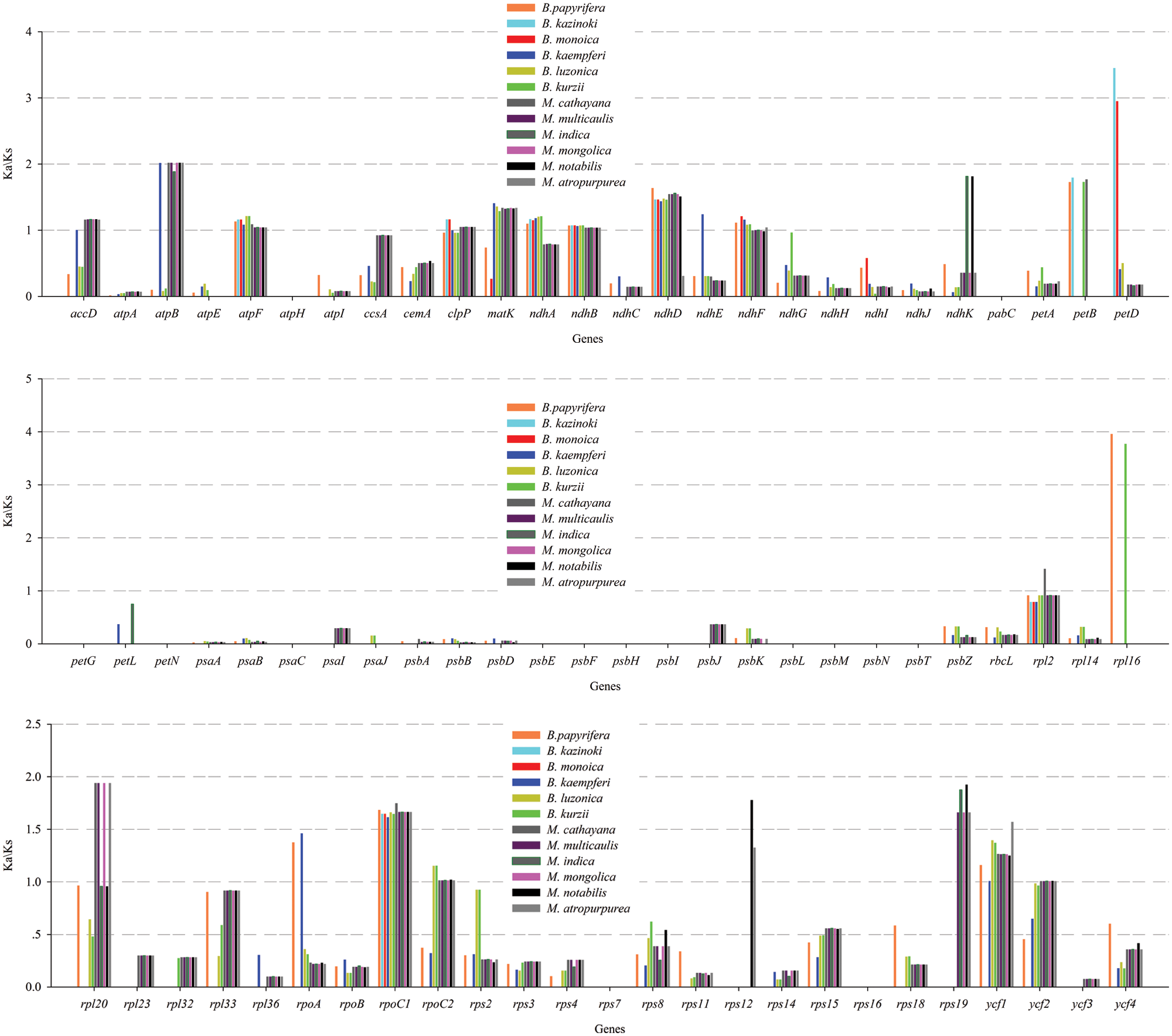
Figure 6: The Ka/Ks ratios of 77 shared PCGs in 12 Moraceae cp genomes were compared with ZJGS.
In the phylogenetic tree based on 77 shared PCGs, ZJGS and its maternal B. kazinoki formed a single clade with high bootstrap support (100%) through two different methods (Fig. 7). All species are divided into four branches according to their evolutionary history. The first branch is the family of Moraceae, which contains genus of Broussonetia, Ficus and Morus. Here, Broussonetia and Ficus are highly clustered with bootstrap values of 100%. In addition, ZJGS, B. kazinoki and B. monoica formed a well-supported clade, as a natural hybrid, B. kazinoki also has the closest phylogenetic relationship with its maternal B. monoica. The second branch, Cannabaceae, includes C. sativa and H. lupulus, which is closely to Moraceae. Studies have shown that both Cannabaceae and Moraceae belong to the Urticalean Rosid clade (Leme et al., 2020; Sytsma et al., 2002). The remaining two families, Rosaceae and Fagaceae, belong to Rosanae. In general, the classification results provide important information for the phylogenetic of ZJGS, as well as confirm the source of ZJGS hybridization.
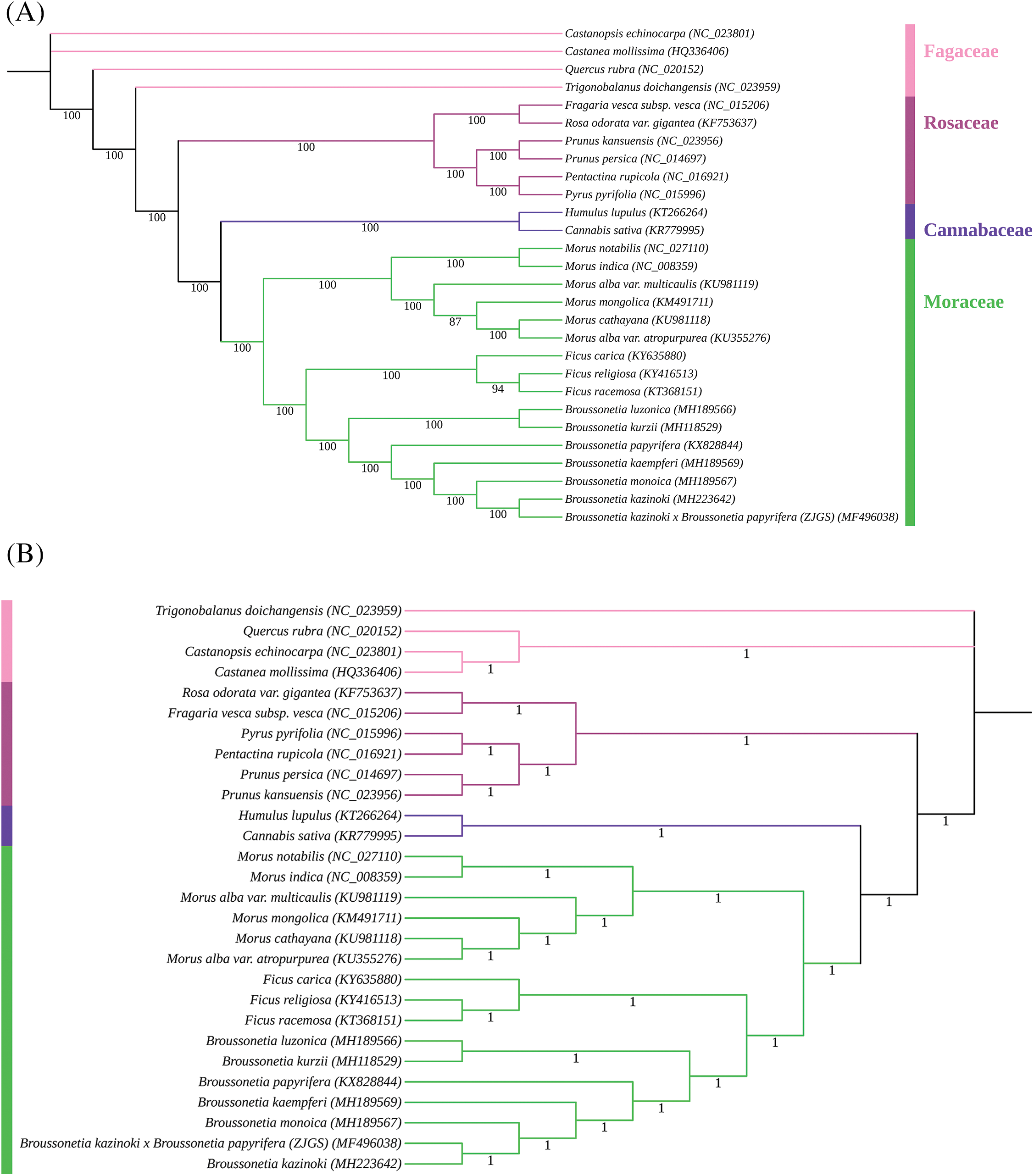
Figure 7: Phylogenetic tree was constructed for ZJGS and 27 related species using different methods based on the 77 shared PCGs. (A) ML phylogenies based on IQ-TREE; (b) BI phylogenies based on the GTR + I + G + F model.
In this study, the cp genome of ZJGS (a hybrid plant) was analyzed with the Moraceae species from structural comparison and sequence alignment. From the structure composition of the cp genome, it can be seen that the seven Broussonetia species and the six Morus species are grouped independently and each of them have similar compositions. Among the seven Broussonetia species, total GC content of ZJGS cp genome sequence is the closest to the maternal B. kazinoki, shows high species affinity (Guo et al., 2020), which is also reflected in the comparison of sequence diversity and SSRs. In addition, the sequence comparison analysis of ZJGS cp genome and other 12 Moraceae plants at the genomic level reflects the high sequence conservation of the IRs regions, so the changes of IRs regions can be used as a marker of species evolution (Huang et al., 2014; Wang et al., 2008). At the boundary of the IRs regions, the changes in the length of the rpl2, rps19, trnN and ycf1 reflect the contraction and expansion of the IRs regions. Through the previous study of sequence diversity, we know that the IRs regions are the most conserved regions in the cp genome. Most studies have shown that the contraction and expansion of the IRs regions determine the length of the cp genome, and this change directly affects the length and distribution of genes at the boundary of this region (Guo et al., 2020; Ivanova et al., 2017; Ravi et al., 2006; Zhang et al., 2013). In the evolution of higher plants, all kinds of repetitive elements have a common feature, which tends to show “co-evolution”, which plays a crucial role in the genome sequence diversity and gene rearrangement (Dover and Coen, 1981; Hanson et al., 1998). Studies have shown that the number of repeats is related to the degree of genome rearrangement (Guisinger et al., 2011; Lee et al., 2007; Xue et al., 2019). Here, long repeat sequences in ZJGS cp genome are the largest, and the number of 20–30 bp repeats may promote the rearrangement of ZJGS (Weng et al., 2014), these repeats can serve as good molecular markers for species evolution, and play an important role in the variation of cp genome sequence.
The breeding process of ZJGS is long and complicated. It has undergone many cross-breeding and space mutations to form excellent plants with stable inheritance. And its maternal B. kazinoki has experienced controversy (Chung et al., 2017; Won, 2019), therefore, it is necessary to compare ZJGS and its hybrid parent systematically. Unlike the previous study (Xu et al., 2018), the cp genome of ZJGS was analyzed in detail. The results of total length and total GC content reflect the high species affinity of ZJGS and maternal species in the phylogenetic history (Choi and Park, 2015; Guo et al., 2020). Similarly, in the sequence diversity comparison, we also found that the cp genome sequence of ZJGS and maternal B. kazinoki remains highly consistence. This is related to sequence conservation and provides a basis for the species evolution of ZJGS (Ivanova et al., 2017; Yin et al., 2018). The construction of the phylogenetic tree shows the most intuitive results, closely related species are grouped into the same branch. And ZJGS is closest to maternal B. kazinoki, which strongly supports the characteristics of cp maternal inheritance (Corriveau and Coleman, 1988; Quan et al., 2003).
Ka/Ks ratio represents the ratio between non-synonymous substitution (Ka) and synonymous substitution (Ks) at a particular site, which is used to infer the direction and magnitude of natural selection acting on PCGs. A ratio >1 means positive selection; <1 means purifying selection; and = 1 indicates no selection (Hurst, 2002; Yang and Bielawski, 2000). According to previous study, except for the most genes with faster evolution rate, the frequency of synonymous nucleotide substitutions is higher than non-synonymous substitutions due to the purification selection (Ivanova et al., 2017). We can screen some genes according to Ka/Ks ratio and then carry out functional studies, which have been commonly applied to the field of molecular evolution (Tae-Kun and Hirohisa, 2008; Yoshihiro et al., 2002). In our research, three genes of atpF, ndhB and rpoC1 show a complete positive selection. In previous studies, some genes have been reported with a faster evolution rate, including ycf1, ycf2, accD, clpP, ndhA, rbcL, matK, ccsA and cemA (Salamin et al., 2013; Stephan et al., 2008). Compared with two cross parents, the same genes (atpF, ndhA, ndhB, ndhD, petB and rpoC1) with positive selection were screened out. Most of the NADH genes (ndhA-ndhK) have a higher Ka/Ks ratio than photosynthesis genes (Photosystem I: psaA, psaB, psaC, psaI, psaJ; Photosystem II: psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbM, psbN, psbT, psbZ), which is similar to the previous research (Choi and Park, 2015; Yang et al., 2016), shows that photosynthesis genes have strong purification selection. NADH genes have higher activity during cellular senescence and oxidative stress (Martín et al., 1996; Peng et al., 2011), and early hybrid selection experiments showed that most proteins synthesized by chloroplasts in the early stages of aging are NDH polypeptides (Vera et al., 1990). It can be seen that these genes under positive selection belong to the advantageous genes of hybrid plants, and may be the key to speciation.
This study is based on the cp genome of ZJGS, a hybrid species, whose structure and composition are the same as most angiosperms, and the IRs regions are highly conserved. The detailed comparison of 13 closely related Moraceae plants revealed the evolutionary characteristics of ZJGS cp genome: (1) The comparison of total GC content, sequence diversity, and SSRs all indicate that the cp genome of ZJGS has significant maternal inheritance; (2) The high sequence variability of the LSC region, the SSC region, and the intergenic regions, and the contraction and expansion of the IRs regions bounder leads to the changes in gene length; (3) Compared with Morus species, the Broussonetia species have a longer cp genome length, higher total GC content and SSRs number. And among all the Broussonetia plants, ZJGS is closest to maternal B. kazinoki. (4) Ka/Ks ratio reveals positive selection of genes such as atpF, ndhB and rpoC1, and NADH genes. Phylogenetic analysis supporting the close relationship between ZJGS and its hybrid parents (the maternal B. kazinoki and the paternal B. papyrifera). And it strongly supports the genetic relationship between ZJGS and maternal B. kazinoki. Our results provide a basis for overcoming phylogenetic problems at the species level, and also provide important genomic resources for the functional utilization of ZJGS.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article (and its supplementary information files). The sequencing data can be downloaded from NCBI Genebank (https://www.ncbi.nlm.nih.gov/).
Author Contribution: The authors confirm contribution to the paper as follows: study conception and design: XZ; data collection: XZ, ZW, YG; analysis and interpretation of results: XZ, ZW, ZJ; draft manuscript preparation: XZ, ZW; funding acquisition, XZ, ZW, HH, ZY. All authors reviewed the results and approved the final version of the manuscript.
Funding Statement: This work was supported by Hunan Provincial Natural Science Foundation of China [Grant No. 2019JJ50027]; China Postdoctoral Science Foundation [Grant No. 2020M683592]; Key Projects of National Forestry and Grassland Bureau [Grant No. 201801]; and Scientific Innovation Fund for Post-graduates of Central South University of Forestry and Technology [Grant No. CX20191004].
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Brudno M, Malde S, Poliakov A, Do CB, Couronne O et al. (2003). Glocal alignment: Finding rearrangements during alignment. Bioinformatics 19: 54–62. [Google Scholar]
Corriveau JL, Coleman AW (1988). Rapid screening method to detect potential biparental inheritance of plastid dna and results for over 200 angiosperm species. American Journal of Botany 75: 1443–1458. [Google Scholar]
Chang CS, Liu HL, Moncada X, Seelenfreund A, Seelenfreund D, Chung KF (2015). A holistic picture of Austronesian migrations revealed by phylogeography of Pacific paper mulberry. Proceedings of the National Academy of Sciences of the United States of America 112: 13537–13542. [Google Scholar]
Choi KS, Park S (2015). The complete chloroplast genome sequence of Aster spathulifolius (Asteraceae); Genomic features and relationship with Asteraceae. Gene 572: 214–221. [Google Scholar]
Chung KF, Kuo WH, Hsu YH, Li YH, Rubite RR et al. (2017). Molecular recircumscription of Broussonetia (Moraceae) and the identity and taxonomic status of B. kaempferi var. australis. Botanical Studies 58: 11. [Google Scholar]
Dover G, Coen E (1981). Springcleaning ribosomal DNA: A model for multigene evolution? Nature 290: 731–732. [Google Scholar]
Gowda M, Kling C, Würschum T, Liu W, Maurer HP et al. (2010). Hybrid breeding in durum wheat: Heterosis and combining ability. Crop Science 50: 2224–2230. [Google Scholar]
Guisinger MM, Kuehl JV, Boore JL, Jansen RK (2011). Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Molecular Biology and Evolution 28: 583–600. [Google Scholar]
Guo L, Guo S, Xu J, He L, Carlson JE, Hou X (2020). Phylogenetic analysis based on chloroplast genome uncover evolutionary relationship of all the nine species and six cultivars of tree peony. Industrial Crops and Products 153: 112567. [Google Scholar]
Guo M, Rupe MA, Dieter JA, Zou J, Spielbauer D et al. (2010). Cell number regulator1 affects plant and organ size in maize: Implications for crop yield enhancement and heterosis. Plant Cell 22: 1057–1073. [Google Scholar]
Hanson RE, Zhao XP, Islam-Faridi MN, Paterson AH, Zwick MS et al. (1998). Evolution of interspersed repetitive elements in Gossypium (Malvaceae). American Journal of Botany 85: 1364–1368. [Google Scholar]
Hori K, Tono A, Fujimoto K, Kato J, Ebihara A et al. (2014). Reticulate evolution in the apogamous Dryopteris varia complex (Dryopteridaceae, subg. Erythrovariae, sect. Variae) and its related sexual species in Japan. Journal of Plant Research 127: 661–684. [Google Scholar]
Huang H, Shi C, Liu Y, Mao SY, Gao LZ (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evolutionary Biology 14: 151. [Google Scholar]
Huang X, Yang S, Gong J, Zhao Y, Feng Q et al. (2015). Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nature Communications 6: 6258. [Google Scholar]
Hurst LD (2002). The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends in Genetics 18: 486–487. [Google Scholar]
Ivanova Z, Sablok G, Daskalova E, Zahmanova G, Apostolova E et al. (2017). Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Frontiers in Plant Science 8: 204. [Google Scholar]
Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proceedings of the National Academy of Sciences of the United States of America 104: 19369–19374. [Google Scholar]
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nature Methods 14: 587–589. [Google Scholar]
Katoh K, Standley DM (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [Google Scholar]
Kobliha J, Stejskal J (2009). Recent fir hybridization research in the light of Czech-American cooperation. Journal of Forest Science 55: 162–170. [Google Scholar]
Kobliha J, Stejskal J, Lstibůrek M, Typta J, Jakubův P (2013). Testing of hybrid progenies and various species of genus Abies for forestry, decorating horticulture and christmas tree production. Acta Scientiarum Polonorum-Hortorum Cultus 12: 85–94. [Google Scholar]
Kong WQ, Yang JH (2017). The complete chloroplast genome sequence of Morus cathayana and Morus multicaulis, and comparative analysis within genus Morus L. PeerJ 5: e3037. [Google Scholar]
Kumar S, Stecher G, Tamura K (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874. [Google Scholar]
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J et al. (2001). REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Research 29: 4633–4642. [Google Scholar]
Lee HL, Jansen RK, Chumley TW, Kim KJ (2007). Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Molecular Biology and Evolution 24: 1161–1180. [Google Scholar]
Leme FM, Borella PH, Marinho CR, Teixeira SP (2020). Expanding the laticifer knowledge in Cannabaceae: Distribution, morphology, origin, and latex composition. Protoplasma 257: 1183–1199. [Google Scholar]
Li D, Qi X, Li X, Li L, Zhong C, Huang H (2013). Maternal inheritance of mitochondrial genomes and complex inheritance of chloroplast genomes in Actinidia Lind.: Evidences from interspecific crosses. Molecular Genetics and Genomics 288: 101–110. [Google Scholar]
Librado P, Rozas J (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [Google Scholar]
Liesebach M, Wuehlisch G, Muhs HJ (1999). Aspen for short-rotation coppice plantations on agricultural sites in Germany: Effects of spacing and rotation time on growth and biomass production of aspen progenies. Forest Ecology & Management 121: 25–39. [Google Scholar]
Lin CS, Chen JJW, Chiu CC, Hsiao HCW, Yang CJ et al. (2017). Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant Journal 90: 994–1006. [Google Scholar]
Lin T, Zhou C, Chen G, Yu J, Wu W et al. (2020). Heterosis-associated genes confer high yield in super hybrid rice. Theoretical and Applied Genetics 133: 3287–3297. [Google Scholar]
Martín M, Casano LM, Sabater B (1996). Identification of the product of ndhA gene as a thylakoid protein synthesized in response to photooxidative treatment. Plant & Cell Physiology 37: 293. [Google Scholar]
Moncada X, Payacán C, Arriaza F, Lobos S, Seelenfreund D et al. (2013). DNA extraction and amplification from contemporary polynesian bark-cloth. PLoS One 8: e56549. [Google Scholar]
Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proceedings of the National Academy of Sciences of the United States of America 107: 4623–4628. [Google Scholar]
Morgan EC, Overholt WA, Sellers Brent (2016). Wildland weeds: Paper mulberry, Broussonetiapapyrifera. University of Florida, IFAS Extension: ENY-702. https://edis.ifas.ufl.edu/publication/in498. [Google Scholar]
Nadachowska-Brzyska K, Li C, Smeds L, Zhang G, Ellegren H (2015). Temporal dynamics of avian populations during Pleistocene revealed by whole-genome sequences. Current Biology 25: 1375–1380. [Google Scholar]
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015a). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution 32: 268–274. [Google Scholar]
Nguyen PAT, Kim JS, Kim J (2015b). The complete chloroplast genome of colchicine plants (Colchicum autumnale L. and Gloriosa superba L.) and its application for identifying the genus. Planta 242: 223–237. [Google Scholar]
Ni J, Su S, Li H, Geng Y, Zhou H et al. (2020). Distinct physiological and transcriptional responses of leaves of paper mulberry (Broussonetia kazinoki × B. papyrifera) under different nitrogen supply levels. Tree Physiology 40: 667–682. [Google Scholar]
Peng L, Yamamoto H, Shikanai T (2011). Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1807: 945–953. [Google Scholar]
Peng X, Teng L, Wang X, Wang Y, Shen S (2014). De novo assembly of expressed transcripts and global transcriptomic analysis from seedlings of the paper mulberry (Broussonetia kazinoki × Broussonetia papyifera). PLoS One 9: e97487. [Google Scholar]
Peng X, Wu Q, Teng L, Tang F, Pi Z, Shen S (2015). Transcriptional regulation of the paper mulberry under cold stress as revealed by a comprehensive analysis of transcription factors. BMC Plant Biology 15: 108. [Google Scholar]
Peng XJ, Liu H, Chen PL, Tang F, Hu YM et al. (2019). A chromosome-scale genome assembly of paper mulberry (Broussonetia papyrifera) provides new insights into its forage and papermaking usage. Molecular Plant 12: 661–677. [Google Scholar]
Powell W, Machray GC, Provan J (1996). Polymorphism revealed by simple sequence repeats. Trends in Plant Science 1: 215–222. [Google Scholar]
Pucher A, Sy O, Sanogo MD, Angarawai II, Zangre R et al. (2016). Combining ability patterns among West African pearl millet landraces and prospects for pearl millet hybrid breeding. Field Crops Research 195: 9–20. [Google Scholar]
Quan Z, Yang L, Sodmergen (2003). Examination of the cytoplasmic DNA in male reproductive cells to determine the potential for cytoplasmic inheritance in 295 angiosperm species. Plant & Cell Physiology 44: 941–951. [Google Scholar]
Ravi V, Khurana JP, Tyagi AK, Khurana P (2006). The chloroplast genome of mulberry: Complete nucleotide sequence, gene organization and comparative analysis. Tree Genetics & Genomes 3: 49–59. [Google Scholar]
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A et al. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biologists 61: 539–542. [Google Scholar]
Rytter L (2002). Nutrient content in stems of hybrid aspen as affected by tree age and tree size, and nutrient removal with harvest. Biomass and Bioenergy 23: 13–25. [Google Scholar]
Rytter L, Stener LG (2005). Productivity and thinning effects in hybrid aspen (Populus tremula L. × P. tremuloides Michx.) stands in southern Sweden. Forestry: An International Journal of Forest Research 78: 285–295. [Google Scholar]
Ryu HW, Curtis-Long MJ, Jung S, Jeong IY, Kim DS et al. (2012). Anticholinesterase potential of flavonols from paper mulberry (Broussonetia papyrifera) and their kinetic studies. Food Chemistry 132: 1244–1250. [Google Scholar]
Salamin N, Dong W, Xu C, Cheng T, Zhou S (2013). Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS One 8: e77965. [Google Scholar]
Stephan G, Xi W, Herrmann RG, Uwe R, Klaus M et al. (2008). The complete nucleotide sequences of the 5 genetically distinct plastid genomes of Oenothera, subsection Oenothera: II. A microevolutionary view using bioinformatics and formal genetic data. Molecular Biology and Evolution 25: 2019–2030. [Google Scholar]
Sugiura M (2015). The gene for chloroplast ribosomal protein S16 is located in both chloroplast and nuclear genomes: Expression of its chloroplast gene. Plant & Animal Genome: P0223. https://pag.confex.com/pag/xxiii/webprogram/Paper17381.html. [Google Scholar]
Suo Z, Li W, Jin X, Zhang H (2016). A new nuclear DNA marker revealing both microsatellite variations and single nucleotide polymorphic loci: A case study on classification of cultivars in Lagerstroemia indica L. Journal of Microbial & Biochemical Technology 8: 266–271. [Google Scholar]
Sytsma KJ, Morawetz J, Pires JC, Nepokroeff M, Conti E et al. (2002). Urticalean rosids: Circumscription, rosid ancestry, and phylogenetics based on rbcL, trnL-F, and ndhF sequences. American Journal of Botany 89: 1531–1546. [Google Scholar]
Tae-Kun S, Hirohisa K (2008). Synonymous substitutions substantially improve evolutionary inference from highly diverged proteins. Systematic Biology 57: 367–377. [Google Scholar]
Timmis JN, Ayliffe MA, Huang CY, Martin W (2004). Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nature Reviews Genetics 5: 123–135. [Google Scholar]
Tompkins DM, Mitchell RA, Bryant DM (2006). Hybridizatfigion increases measures of innate and cell-mediated immunity in an endangered bird species. Journal of Animal Ecology 75: 559–564. [Google Scholar]
Umadevi M, Veerabadhiran P, Manonmani S (2014). Assessment of genetic diversity of rice (Oryza sativa) cultivars using ssr markers. African Journal of Biotechnology 13: 3547–3552. [Google Scholar]
Vera A, Tomás R, Martín M, Sabater B (1990). Apparent expression of small single copy cpDNA region in senescent chloroplasts. Plant Science 72: 63–67. [Google Scholar]
Wang RJ, Cheng CL, Chang CC, Chaw SM (2008). Dynamics and evolution of the inverted repeat/large single copy junctions in the chloroplast genomes of monocots. BMC Evolutionary Biology 8: 36. [Google Scholar]
Weng ML, Blazier JC, Govindu M, Jansen RK (2014). Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Molecular Biology and Evolution 31: 645–659. [Google Scholar]
Won H (2019). Test of the hybrid origin of Broussonetia × kazinoki (Moraceae) in Korea using molecular markers. Korean Journal of Plant Taxonomy 49: 282–293. [Google Scholar]
Xu ZG, Yang GY, Dong M, Wu L, Zhang W, Zhao YL (2018). The complete chloroplast genome of an economic and ecological plant, paper mulberry (Broussonetia kazinoki × Broussonetia papyifera). Mitochondrial DNA Part B 3: 28–29. [Google Scholar]
Xue S, Shi T, Luo W, Ni X, Iqbal S et al. (2019). Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Horticulture Research 6: 89. [Google Scholar]
Yan J, Wu PS, Du HZ, Zhang QE (2011). First report of black spot caused by Colletotrichum gloeosporioides on paper mulberry in China. Plant Disease 95: 880. [Google Scholar]
Yang Y, Tao Z, Dong D, Jia Y, Li F, Zhao G (2016). Comparative analysis of the complete chloroplast genomes of five Quercus species. Frontiers in Plant Science 7: 959. [Google Scholar]
Yang ZH, Bielawski JP (2000). Statistical methods for detecting molecular adaptation. Trends in Ecology & Evolution 15: 496–503. [Google Scholar]
Yin K, Zhang Y, Li Y, Du FK (2018). Different natural selection pressures on the atpF gene in evergreen sclerophyllous and deciduous oak species: evidence from comparative analysis of the complete chloroplast genome of Quercus aquifolioides with other oak species. International Journal of Molecular Sciences 19: 1042. [Google Scholar]
Yoshihiro M, Yukiko Y, Yasunari O, Koichiro T (2002). Whole chloroplast genome comparison of rice, maize, and wheat: implications for chloroplast gene diversification and phylogeny of cereals. Molecular Biology and Evolution 19: 2084–2091. [Google Scholar]
Zhang D, Gao F, Li WX, Jakovlić I, Zou H et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Molecular Ecology Resources 20: 348–355. [Google Scholar]
Zhang H, Li C, Miao H, Xiong S (2013). Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS One 8: e80508. [Google Scholar]
Zhao XP, Liu J, Xia X, Chu J, Wei Y et al. (2014). The evaluation of heavy metal accumulation and application of a comprehensive bio-concentration index for woody species on contaminated sites in Hunan, China. Environmental Science and Pollution Research 21: 5076–5085. [Google Scholar]
Supplementary files
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |