DOI:10.32604/biocell.2021.016500
| BIOCELL DOI:10.32604/biocell.2021.016500 | |
| Article |
Characterization of endogenous nucleic acids that bind to NgAgo in Natronobacterium gregoryi sp2 cells
1School of Life Science and Technology, University of Electronic Science and Technology of China, Chengdu, 611731, China
2School of Healthcare Technology, Chengdu Neusoft University, Chengdu, 611844, China
3School of Medicine, Guizhou University, Guiyang, 550025, China
*Address correspondence to: Jian Huang, hj@uestc.edu.cn
Received: 11 March 2021; Accepted: 16 April 2021
Abstract: As nucleic acid-guided endonucleases, some prokaryotic Argonautes have been used as programmable nucleases. Natronobacterium gregoryi Argonaute (NgAgo) has also been proposed for gene editing, but this remains very controversial. Until now, the endogenous nucleic acids that bind to NgAgo in Natronobacterium gregoryi sp2 (N. gregoryi sp2) have not been characterized. We expressed the conserved PIWI domain of NgAgo and used it to induce anti-PIWI antibody. We also cultured the N. gregoryi sp2 strain and performed immunoprecipitation, chromatin immunoprecipitation (ChIP), and RNA immunoprecipitation (RIP) assays. The nucleic acids that endogenously bound NgAgo in N. gregoryi sp2 cells were sequenced and analyzed. The results showed that NgAgo endogenously bound RNA rather than DNA. NgAgo-associated RNAs were mainly transcripts of genes that encoded tRNA, transcriptional regulators, RNA polymerases, and RNA-binding proteins. NgAgo mainly binds to the transcripts inside genes or in their upstream sequences. Interestingly, the top enriched motif of peaks was the same as that of miR-1289, suggesting that NgAgo may regulate gene expression post-transcriptionally. GO enrichment analysis showed that the peak-associated genes were enriched in transmembrane transport processes. These results revealed that NgAgo binds RNA and may function in post-transcriptional regulation in vivo.
Keywords: NgAgo; PIWI; Immunoprecipitation; ChIP-seq; RIP-seq; Post-transcriptional regulation
Eukaryotic Argonautes (eAgos) bind small RNAs and use them as guides for sequence-specific recognition of long RNA targets in a process known as RNA interference (RNAi) (Niaz, 2018). By contrast, many prokaryotic Agos (pAgos) contain divergent variants of conserved nucleic acid interaction domains, as well as extra domains that are absent from eAgos. This suggests that pAgos may have unusual specificities in nucleic acid recognition and transcription inhibition or cleavage (Ryazansky et al., 2018). Agos contain four domains that are organized in a bilobal structure and two linker domains (L1 and L2). The N-terminal and PAZ (PIWI-Argonaute-Zwille) domains form one lobe, and the MID (middle) and PIWI (P-element Induced Wimpy Testis) domains form the other lobe. Nucleic acids are bound between the lobes. The MID and PAZ domains interact with the 5’ and 3’ ends of small nucleic acid guides. The PIWI domain contains an RNase H-like fold with a catalytic tetrad of conserved amino acid residues. The N-terminal domain is the least conserved portion of the Ago protein, in some pAgos, it is proposed to prevent extended duplex formation during target recognition (Hur et al., 2014; Lisitskaya et al., 2018; Olina et al., 2018; Ryazansky et al., 2018; Swarts et al., 2014). AGONOTES (Jiang et al., 2020) was developed to identify Agos and annotate their domains based on proteome or protein sequences.
Many pAgos associated with putative nucleases, helicases, and DNA binding proteins within the same gene or operon have been proposed as potential tools for genome engineering (Swarts et al., 2014). Thermus thermophilus Argonaute (TtAgo)-gDNA has been demonstrated to cleave RNA targets in vitro (Lei et al., 2019; Swarts et al., 2015; Swarts et al., 2017; Zhu et al., 2016). The archaeon Pyrococcus furiosus Argonaute (PfAgo) and Methanocaldococcus jannaschii Argonaute (MjAgo) mediate DNA-guided DNA cleavage in vitro (Hegge et al., 2018). Archaeoglobus fulgidus Argonaute (AfAgo) has a preference for an ssDNA guide over an ssRNA guide for binding DNA targets in vitro (Ma et al., 2005; Parker et al., 2005). Genes encoding Marinitoga piezophila Argonaute (MpAgo) and Thermotoga profunda Argonaute (TpAgo) cluster with genes encoding CRISPR Cas enzymes, suggesting a functional link. MpAgo and TpAgo use 5’-hydroxyl group gRNA to cleave cognate ssDNA targets in vitro. MpAgo can also use a 5’-hydroxylated RNA guide to cleave ssRNA in vitro (Kaya et al., 2016). Aquifex aeolicus Argonaute (AaAgo) mediates DNA-guided RNA cleavage in vitro (Yuan et al., 2005). DNA-guided DNA cleavage by Clostridium butyricum Argonaute (CbAgo) and Limnothrix rosea Argonaute (LrAgo) has been observed in vitro (Hegge et al., 2019; Kuzmenko et al., 2019). Additionally, TtAgo-gDNA targets invading nucleic acids and cleaves single- and double-stranded DNA in vivo (Swarts et al., 2015). PfAgo was demonstrated to decrease the efficiency of plasmid transformation in P. furiosus in vivo (Hegge et al., 2018). Rhodobacter sphaeroides Argonaute (RsAgo)-RNA/DNA represses the expression of plasmids transcriptionally and/or post-transcriptionally by an unknown mechanism in vivo (Olovnikov et al., 2013). Many pAgos associated with DNA and/or RNA in vivo still remain uncharacterized, such as Methanopyrus kandleri Argonaute (MkAgo) and NgAgo.
NgAgo-gDNA was previously reported to cleave dsDNA at 37°C (Gao et al., 2016). This article was later retracted because the results could not be reproduced by many laboratories (Cyranoski, 2016; Javidi-Parsijani et al., 2017; Lee et al., 2016). A study showed that the NgAgo/gDNA system could knock down the fabp11a gene in zebrafish, and it was suggested that this occurred through binding to the fabp11a coding sequence (Qi et al., 2016). However, another study suggested that NgAgo instead mediated DNA-guided RNA cleavage. It has been demonstrated that NgAgo/gDNA regulates gene expression post-transcriptionally in vitro in a manner that resembles eukaryotic RNAi pathways (Sunghyeok, 2017). In addition, the NgAgo-gDNA system was also reported to inhibit HBV replication by accelerating pgRNA degradation (Wu et al., 2017). However, NgAgo was reported to show no meaningful binding to chromosomal targets in mammalian cells (O’geen et al., 2018). Recently, it was reported that NgAgo improved the efficiency of bacterial gene knockout by enhancing recA-mediated homologous DNA strand exchange independent of its DNase activity and exogenous gDNA (Fu et al., 2019). To date, although short DNA has always been used as a guide in all NgAgo-related studies, there have been no relevant studies on the natural nucleic acids that bind to NgAgo in vivo and information that is critical for its characterization. Nevertheless, the preference of NgAgo for DNA binding has not been reported. In this paper, we aim to characterize the nucleic acids that bind endogenously to NgAgo in N. gregoryi sp2 cells.
PIWI expression and purification
The pBBR-Tac-NgAgo plasmid was kindly provided by Chengdu Renhao Biological Technology Co., Ltd., China Recombinant plasmid construction and PIWI domain expression were performed by Hangzhou HuaAn Biotechnology Co., Ltd., China. The 6xHis-Flag-tagged PIWI domain (approximately 38 kDa) of the NgAgo gene was amplified by primers PIWI-F 5’CGGGATCCGCGGCATTCGTCGTACTG3’ and PIWI-R 5’CGCTCGAGGAGGTAACCCTTGGTCGC3’ and cloned into the pET-28a expression vector at the BamHI and XhoI sites (kanamycin-resistant). A recombinant plasmid circle map was drawn with SnapGene software (Insightful Science, snapgene.com) (Fig. 1). The cloned sequence was verified by sequencing. The pET-28a-PIWI plasmid (0.5 μL) was transfected into 100 μL of Rosetta Escherichia coli incubated in 300 mL of liquid LB medium at 37°C for 1 h. PIWI was induced using 1 mM IPTG (Shanghai Suobio Bioscience & Technology Co., Ltd., China.) at 37°C for 4 h. Rosetta cells were lysed by sonication. PIWI was expressed in the inclusion body, and the inclusion body was dialyzed with glycerol, arginine, and glycine gradient buffers (6, 4, 3, 2, 1, and 0 M urea) and dissolved using 1 × PBS. PIWI was eluted from Ni-NTA resin using 5 mL denaturing elution buffer (20 mM sodium phosphate [pH 7.8], 0.5 M sodium chloride, 8 M urea, 300 mM imidazole). The purity of PIWI was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE). The concentrated PIWI was stored at 4°C.
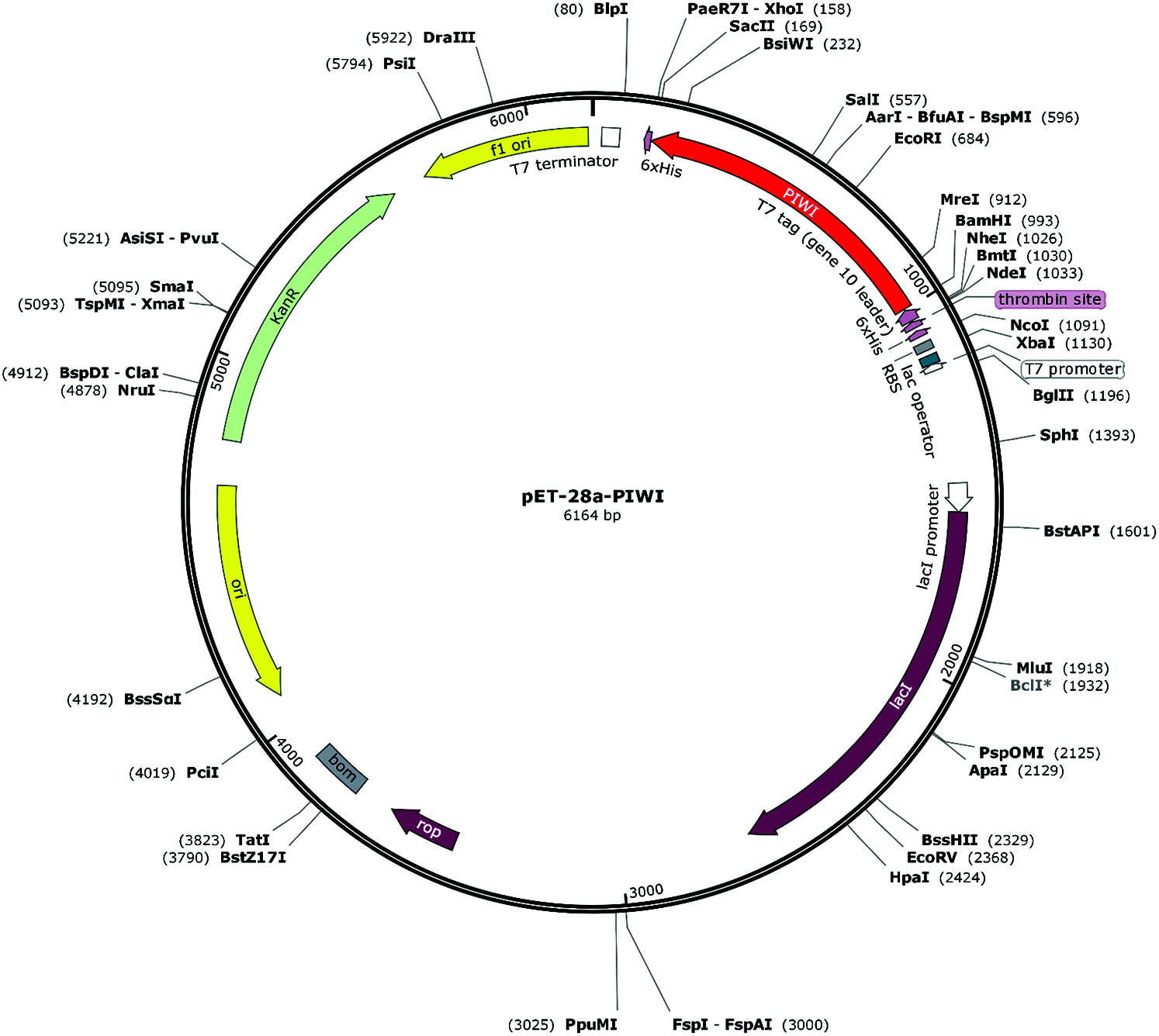
Figure 1: Schematic diagram of the recombinant plasmid pET-28a-PIWI. PIWI was cloned into plasmid pET-28a at the XhoI and BamHI sites (kanamycin-resistant).
Preparation of anti-PIWI polyclonal antibody
Polyclonal antibody serum was prepared by Hangzhou HuaAn Biotechnology Co., Ltd., China. New Zealand white rabbits (~2.5 kg) were subcutaneously injected with 0.2 mL PIWI emulsified with isopycnic Freund’s complete adjuvant. The titer and specificity of the antiserum against PIWI were determined by enzyme-linked immunosorbent assay (ELISA) and western blotting, respectively. Anti-PIWI polyclonal antibody was purified from antiserum by PIWI antigen affinity chromatography. Fifteen milliliters of antiserum were diluted to 20 mL with 50 mM PBS (pH 7.4) and then added to the chromatography column with PIWI antigen. The antibody was eluted twice using 0.2 M glycine-HCl buffer at pH 3.0.
Antiserum was collected after four rounds of immunization, and the anti-PIWI polyclonal antibody titer was determined by ELISA after antibody purification using protein A. Ninety-six-well Falcon immunoplates were coated with 50 μL of PIWI domain (diluted to 1 μg/mL using coating buffer [Na2CO3 and NaHCO3]) overnight at 4°C and blocked with 60 μL 1% bovine serum albumin (BSA) at 37°C for 1 h. Fifty microliters of purified anti-PIWI antibody (1:250, 1:1000, 1:4000, 1:16,000, 1:64,000, 1:256,000 gradient dilution) were added to the plates and incubated at 37°C for 1 h; the plates were then washed twice with TBST. Goat anti-rabbit IgG conjugated with horseradish peroxidase (HRP) was added to the plates at a dilution of 1:5000 as a secondary antibody and signal, and the plates were incubated at 37°C for 45 min. One hundred microliters of TMB chromogenic solution consisting of equal volumes of solution A (disodium hydrogen phosphate, citric acid, hydrogen peroxide, and deionized water) and solution B (TMB, citric acid, EDTA, glycerin, DMSO, and deionized water) were added and allowed to react for 5 min at 37°C. A stop solution (90 μL 2 M sulfuric acid) was added, and the absorbance was measured at 450 nm.
For western blotting of anti-PIWI polyclonal antibody serum, N. gregoryi sp2 cell lysates were mixed with an equal volume of SDS–PAGE loading buffer and boiled at 100°C for 5 min, then immediately put in ice for 10 min. N. gregoryi sp2 cell lysates were then separated on an 8% SDS–PAGE gel at 150 V for 90 min and transferred to a polyvinylidene difluoride (PVDF) membrane at 75 V for 60 min. Next, the membrane was blocked in 5% skim milk buffer in PBST for 1 h at room temperature. The membrane was then incubated with 1 mL antibody solutions (antiserum samples diluted 1:1000 with 5% skim milk buffer) for 1 h at room temperature. This was followed by a reaction with goat anti-rabbit IgG conjugated with HRP as a secondary antibody and signal (diluted 1:5000 with 5% skimmed milk powder buffer) at room temperature for 1 h. After three washing rounds, strips were incubated for 3 min at room temperature with a 1:1 mixture of solution A and solution B.
Archaea strain N. gregoryi sp2 cell culture
N. gregoryi sp2 strain ATCC43098 was purchased from the China General Microbiological Culture Collection Center (CGMCCC) (#1.1967). Cells were grown in 211 minimal medium A at 37°C and 120 rpm under aerobic conditions at pH 9.5 for 5 days. The 211 minimal medium A was prepared using 7.5 g casamino acids, 10.0 g yeast extract, 3.0 g sodium citrate, 0.1 g MgSO4·7H2O, 2.0 g KCl, microscale Fe2+ and Mn2+, 200.0 g NaCl, and 8.0 g anhydrous Na2CO3 (sterilized independently); distilled water was added to give a volume of 1.0 L and a final pH of 9.5.
Protein complex immunoprecipitation and analysis
Immunoprecipitation (IP) assays were performed by Huan Biotechnology Co., Ltd. (Hangzhou, China). N. gregoryi sp2 cells were harvested and solubilized in IP buffer (1% Triton X-100, 1% glycerol, and protease inhibitors [Roche] in PBS). Five hundred micrograms of cell lysate were incubated with a complex of protein A-agarose beads (Santa Cruz Biotechnology) and anti-PIWI or rabbit IgG antibody for 3–5 h at 4°C. Beads were then washed with IP buffer three times and subjected to immunoblotting. Proteins from lysate alone and proteins from the lysate incubated with IgG were used as positive and negative controls, respectively. The anti-PIWI antibody was diluted to 1:50 and reacted with goat anti-rabbit IgG conjugated with HRP as a secondary antibody.
Nucleic acids were extracted from the NgAgo complex and quantified using a Qubit fluorometer and an Agilent 2100 Bioanalyzer (Agilent Technologies) before being prepared for sequencing. RNA read quality was assessed using FastQC, and reads were filtered and trimmed with FASTX-Toolkit to remove adapters, rRNA, and low-quality reads. Trimmed reads were mapped to the N. gregoryi sp2 reference genome using TopHat (version 2.0.9) (Trapnell et al., 2009). RNA transcripts from the NgAgo complex were quantified by fragments per kilobase of exon per million fragments mapped (FPKM) values using Cufflinks (version 2.1.1) (Trapnell et al., 2010).
Isolation of endogenous nucleic acid targets of NgAgo by ChIP and RIP assays
DNA fragments of the N. gregoryi sp2 genome associated with NgAgo were extracted and sequenced by ChIP and ChIP-seq, respectively. Likewise, RNA fragments from N. gregoryi sp2 genome-wide RNA transcripts that interacted with NgAgo were isolated and sequenced by RIP and RIP-seq. ChIP and RIP assays were performed by Zoonbio Biotechnology Co., Ltd., Nanjing, China, and ChIP-seq and RIP-seq were performed by Shanghai Biotechnology Corporation Co., Ltd., China.
The ChIP assay was performed as follows. N. gregoryi sp2 cells were grown in 211 minimal medium A at 37°C. When the cells reached 80% confluency, a 36.5% formaldehyde solution was added to the cell suspension to a final concentration of 1% and incubated for 15 min at room temperature. Up to 0.125 M glycine was added, and the mixture was incubated for 5 min at room temperature to stop the cross-linking reaction. Cells were washed twice with cold PBS containing protease inhibitor cocktail, and the cross-linked cells were centrifuged for 5 min at 4°C. The cells were harvested and resuspended in 300 μL 1% SDS lysis buffer containing protease inhibitor cocktail for 30 min on ice to ensure cell lysis. The cell lysates were sonicated to shear cellular DNA and centrifuged for 10 min at 4°C to remove cellular debris. Electrophoresis on a 1% agarose gel indicated that DNA fragment size ranged from 60 to 500 bp. Ten microliters of chromatin solution containing the total genomic DNA fraction were saved at −20°C for later use. Anti-PIWI antibody (5 μL) was added to the chromatin solution and incubated overnight at 4°C, and a tube with rabbit IgG was included as a negative control. The agarose bead antibody-chromatin complexes were centrifuged at 500 × g for 4 min at 4°C, and the beads were washed with wash buffers. The immune complexes were eluted from the agarose beads using 250 μL of elution buffer and incubated for 30 min at room temperature. Twenty microliters of 5 M NaCl were added to each tube, and cross-linking was reversed by heating at 65°C from 4 h to overnight. Ten microliters of 0.5 M EDTA, 20 μL of 1 M Tris–HCl (pH 6.5), and 2 μL of proteinase K (10 mg/mL) were added to each tube, incubated for 2 h at 45°C, and treated with RNase A (10 μg/μL) at 37°C for 2 h. To recover DNA, 500 μL of phenol/chloroform/isoamyl alcohol was added to each tube; tubes were shaken thoroughly for 15 s, and the supernatant was collected after centrifuging the samples at 13,000 × g for 15 min at 4°C. The DNA was extracted again using phenol/chloroform/isoamyl alcohol, and the supernatant was transferred to a 2-mL tube. To each tube were added 2.5 volumes of absolute ethanol, 1/10 volume of 3 M sodium acetate (pH 5.2), and 1 μL of glycogen (20 mg/mL). Tubes were incubated from 4 h to overnight at −20°C to precipitate DNA. DNA was dissolved in 50 μL of TE and stored at −20°C until analysis. Washing, crosslink reversal, and purification of the ChIP DNA were performed, and all DNA samples were quantified using a NanoDrop 2000 spectrophotometer.
Native RIP assays (Gagliardi and Matarazzo, 2016) were performed using an RIP assay kit (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. All buffers and solutions were free of RNase contamination. For each immunoprecipitation, N. gregoryi sp2 cells were grown to approximately 80% confluence, harvested, and collected by centrifugation. RNA-NgAgo complex lysates (400 μL) were prepared from N. gregoryi sp2 cells using RIP lysis buffer (protease inhibitor cocktail and RNase inhibitor). The lysates were immediately frozen on ice to complete the lysis process. Protein A-sepharose beads were pre-bound with 1 μg of either anti-PIWI antibody or negative control normal rabbit serum IgG antibody using NT-2 buffer (250 mM Tris-HCl [pH 7.4], 750 mM NaCl, 5 mM MgCl2, 0.25% NP-40) at 4°C overnight. Equal amounts of RNA-NgAgo complex lysates (3 mg) were incubated with antibody-precoated sepharose beads. Beads were washed five times with 0.5 mL RIP wash buffer. Each immunoprecipitate was resuspended in 150 μL of proteinase K buffer and incubated at 55°C for 30 min to digest the proteins. All tubes were placed in a magnetic rack, the supernatants were transferred to new tubes, and 250 μL NT-2 was added after 30 min of incubation. Four hundred microliters of phenol:chloroform:isoamyl alcohol (125:24:1) were added to all tubes, and the tubes were centrifuged for 10 min at room temperature to separate the phases. A 350 μL-aliquot of the aqueous phase was placed into a new tube, 400 μL of chloroform was added, and the mixture was centrifuged for 10 min at room temperature to obtain 300 μL of the aqueous phase. Fifty microliters of 5 M ammonium acetate, 15 μL of 7.5 M LiCl, 5 μL glycogen (5 mg/mL), and 850 μL of absolute ethanol were added to each tube, and the tubes were maintained at −80°C overnight.
ChIP-seq libraries were generated for paired-end sequencing using the TruSeq DNA LT Sample Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. In brief, the fragmented DNA samples (1 μg/each, duplicate) were end-repaired, A-tailed at the 3’ end, and ligated with the indexing adapters provided. The potential target DNA samples were extracted using AMPure XP magnetic beads and amplified by PCR to create the final ChIP-seq library. The ChIP-seq library concentration was 12.8 ng/μL quantified with a Qubit 2.0 fluorometer, and the peak length of the library was 256 bp quantified with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA.). The DNA in the ChIP-seq library was sequenced twice with a read length of 150 bp on a HiSeq 2500 instrument (Illumina) according to the manufacturer’s protocol.
For ChIP-seq data analysis, read quality was assessed using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Read filtering and trimming were performed with Trim Galore (version 0.6.5). Reads were aligned to the N. gregoryi sp2 reference genome using Bowtie 2 (version 2.3.5.1) with default parameters (Langmead and Salzberg, 2012). Statistically significantly enriched peaks were identified using MACS2 (version 2.1.1.20160309) (Zhang et al., 2008). Peak annotation and motif enrichment were performed using HOMER (version 4.11) (http://homer.ucsd.edu/homer/motif/) (Heinz et al., 2010).
RNA sample quality was assessed with an Agilent 2100 Bioanalyzer and a Qubit fluorometer. To determine which mRNAs were associated with NgAgo, cDNA library synthesis from NgAgo-mRNA immunocomplexes was performed according to the manufacturer’s protocols (Illumina). In brief, mRNA was purified from 100 ng of immunocomplex total RNA using poly(T) magnetic beads. The mRNA was fragmented into small pieces using divalent cations at an elevated temperature. The RNA fragments were synthesized into first-strand cDNA with reverse transcriptase and random hexamer or oligo(dT) primers, followed by second-strand cDNA synthesis using DNA polymerase I and RNase H. The resulting cDNA fragments were repaired by adding a single “A” base and ligated to adaptors. The ligation products were then purified and amplified by PCR to construct the cDNA library according to the manufacturer’s instructions (Illumina). Finally, the library was analyzed with a Qubit fluorometer to measure concentrations and an Agilent 2100 to measure fragment sizes. The RIP-seq library was sequenced with a read length of 150 bp on a HiSeq 2500 instrument (Illumina) according to the manufacturer’s protocol.
RIP-seq read quality was assessed using FastQC, and the reads were then filtered and trimmed using Trimmomatic (version 0.39) (Bolger et al., 2014). Trimmed RIP-seq reads were mapped to the N. gregoryi sp2 reference genome using TopHat (version 2.0.9) (Trapnell et al., 2009). Expression of NgAgo-binding RNAs was quantified by FPKM values using Cufflinks software (version 2.1.1) (Trapnell et al., 2010). Peak detection and annotation were then performed using RIPSeeker (Li et al., 2013), which is specifically tailored for RIP-seq data analysis. Motif enrichment analysis of peaks was performed with HOMER (version 4.11), and the Gene Ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed to annotate the functions of peaks associated with genes.
NgAgo PIWI production and purification
The His-tagged PIWI domain was heterologously produced and purified. The purification was performed based on a 6X-His tag. PIWI was induced using 1 mM IPTG and dissolved in urea. On SDS–PAGE, PIWI migrated to a position indicative of a mass of approximately 38 kDa (Fig. 2). The PIWI sample and the dialyzed PIWI were both subjected to SDS–PAGE analysis, and PIWI from different samples migrated to the same position of approximately 38 kDa (Fig. 2a lanes 4 and 5, Fig. 2b lanes 2 and 3). Greater amounts of PIWI were eluted with increasing imidazole concentrations within certain limits.
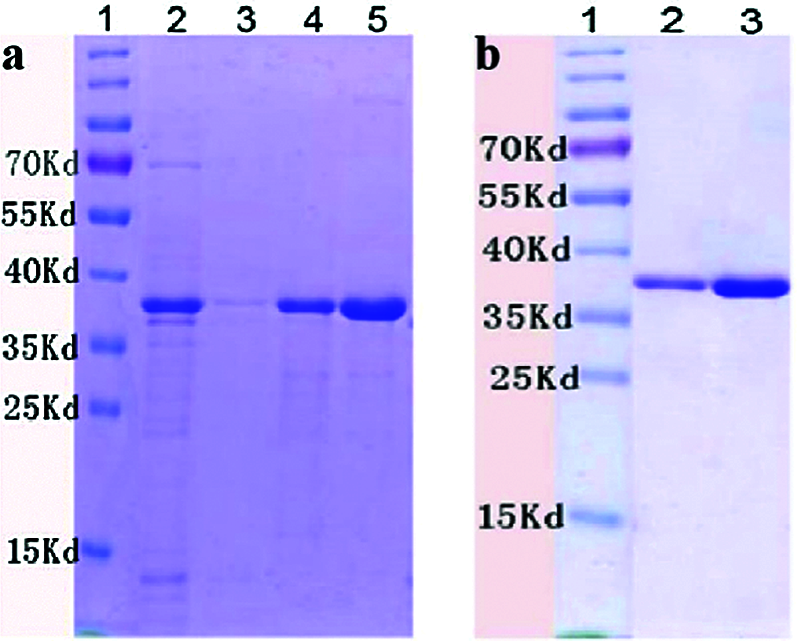
Figure 2: SDS–PAGE of purified PIWI (38 kDa). Conditions for PIWI expression and purification are described in the Methods. The purified PIWI was analyzed by electrophoresis on a 12% SDS gel. (a) Lane 1, molecular weight markers (kDa); lane 2, flow-through liquid; lane 3, washing buffer; lane 4, purified recombinant PIWI (eluted with 30 mM imidazole); lane 5, purified recombinant PIWI (eluted with 300 mM imidazole). (b) PIWI samples were dialyzed with glycerol, arginine, and glycine gradient buffers (6, 4, 3, 2, 1, and 0 M urea). Lane 1, molecular weight markers (kDa). Lanes 2 and 3 correspond to lanes 4 and 5 in (a).
Preparation of anti-PIWI antibody
The anti-PIWI polyclonal antibody titer was determined by ELISA after antibody purification using protein A. The mean value at OD 450 nm of a 1:16,000 dilution of anti-PIWI antibody was 0.737 (>0.6), indicating that the antibody titer was sufficient (Tab. 1). Western blotting was performed with anti-PIWI antibody (1:200 and 1:1000 dilutions) on an 8% SDS gel as described above (Fig. 3a). In the blocked group, the anti-PIWI antibody had been previously incubated with excess PIWI antigen. Therefore, in the blocked group, there were no binding sites on the membrane for PIWI (or NgAgo in the N. gregoryi sp2 cell lysates). The NgAgo band was weaker or absent in the blocked group compared with the experimental group (Fig. 3b). Subsequently, the antibody was purified for western blotting, which again showed a unique band that was bound by purified anti-PIWI antibody (Fig. 3c). The band corresponding to NgAgo and detected by anti-PIWI antibody in N. gregoryi sp2 cell lysates was approximately 120 kDa.
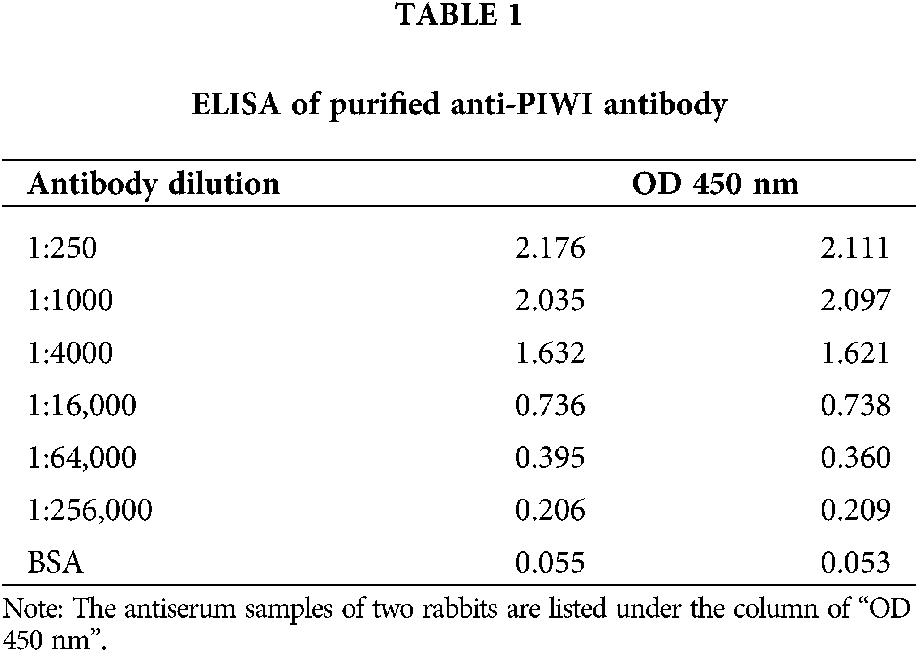
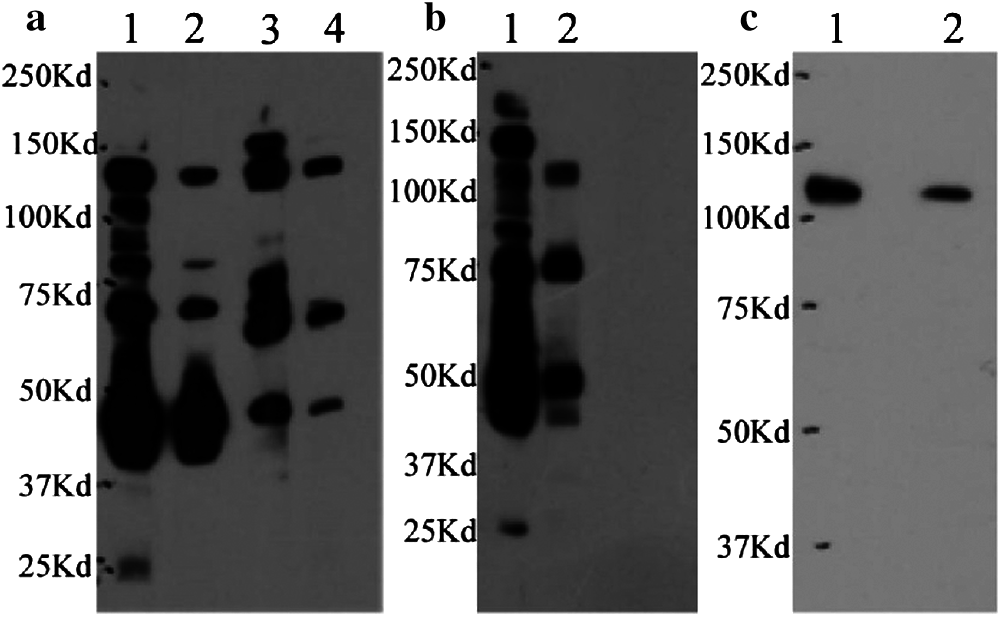
Figure 3: Western blotting for anti-PIWI antibody. (a) Two dilutions of antiserum samples (Lanes 1 and 3 [1:200], Lanes 2 and 4 [1:1000]) from two rabbits (Rabbit 1 in Lanes 1 and 2, Rabbit 2 in Lanes 3 and 4) incubated with N. gregoryi sp2 cell lysates. (b) The anti-PIWI antibody had been previously incubated with excess PIWI antigen in the blocked group (Lane 2). (c) Western blot for purified anti-PIWI antibody (Lane 1 1:50, Lane 2 1:100).
The N. gregoryi sp2 strain is aerobic, Gram-negative, red, and very thick after centrifugation; it grows in an extremely saline environment. The strain was cultured and identified by whole-genome sequencing. Here, the genome of N. gregoryi sp2 was purchased from CGMCCC (Accession Number 1.1967) and assembled by Illumina sequencing. The draft genome sequences have been deposited at GenBank under the accession number PKKI00000000 (Jiang et al., 2019). The draft genome was annotated by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP4.4) with default settings. Conserved PIWI sequences of Argonaute proteins from known bacteria and archaea were used to construct a local BlastP database, and the N. gregoryi sp2 proteome was used as a query. The top five hit sequences with the lowest e-values were selected and annotated using CDART (Geer et al., 2002). Only one sequence was successfully annotated as containing the conserved PIWI domain. The PIWI sequences of MtAgo and TtAgo had the highest similarity with the PIWI of NgAgo. There was only one PIWI domain in N. gregoryi sp2, according to the above analysis.
We performed immunoprecipitation (IP) using an anti-PIWI antibody, extracted nucleic acids from the NgAgo complex and quantified them using a Qubit fluorometer and an Agilent 2100 Bioanalyzer in preparation for sequencing. Only RNA, and no DNA, was detected in the complexes (Fig. 4b). The NgAgo complex band was detected using an anti-PIWI antibody (approximately 120 kDa), as shown in Fig. 4a. The concentration of RNA was 11.8 ng/μL, and the total amount was 0.14 μg.

Figure 4: Western blot of the NgAgo complex from the IP assay and RNA size estimation, quantification, and quality control. (a) Lane 1, positive control, proteins from N. gregoryi sp2 lysate; lane 2, negative control, proteins from the lysate incubated with IgG in the IP assay; lane 3, NgAgo complex from the IP assay. (b) Total RNA was analyzed using an Agilent 2100 Bioanalyzer.
The percentage of rRNA among the raw reads was 98.13%. The proportion of trimmed reads in the raw reads was 75.77%. The trimmed reads that mapped to the genome were 45.58%. The top 20 genes were selected from the RNA-seq gene expression data based on the FPKM value. Analysis of the RNA-seq expression data showed that NgAgo bound significantly to ncRNA (ffs and rnpB), tRNA, and transcripts of non-histone chromosomal MC1 family protein, cold-shock protein, and twin-arginine translocation signal domain-containing protein (Tab. 2). ffs (SRP_RNA) plays a critical role in co-translational protein targeting and delivery to cellular membranes by binding with signal recognition particle (SRP) (Flores and Ataide, 2018; Fukuda et al., 2020). rnpB (RNase_P_RNA) (Kirsebom, 2002; 2007) associates with endoribonuclease P (RNase P) to process tRNA precursors and generate mature 5’ termini. RNase_P_RNA-based catalytic activity has been preserved during evolution in both prokaryotes and eukaryotes.
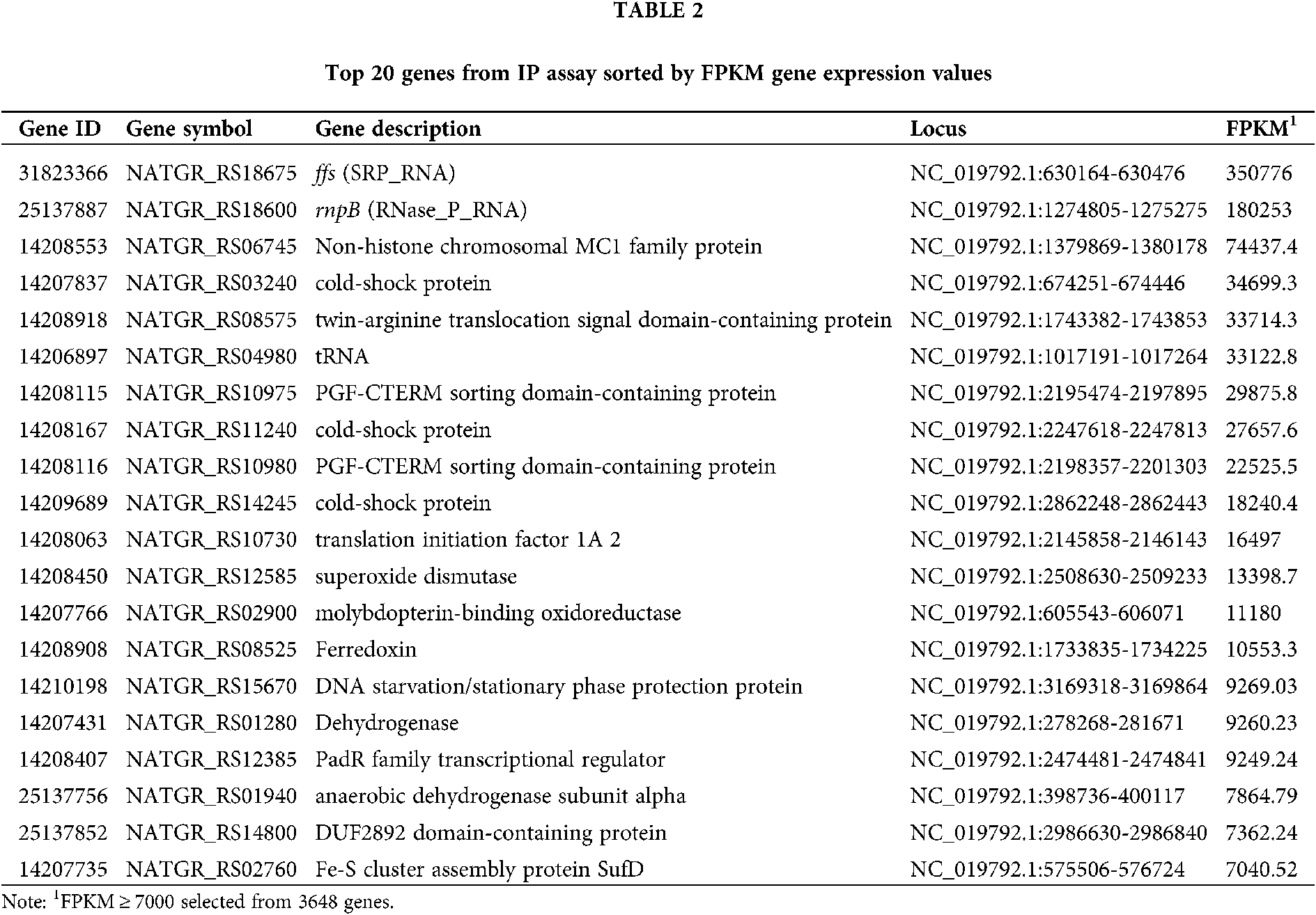
Enrichment of DNA and RNA associated with NgAgo
N. gregoryi sp2 cells were grown in 211 minimal medium A at 37°C for about 5 days. The cells were harvested when they reached approximately 80% confluency for each immunoprecipitation. ChIP and RIP assays were performed using the N. gregoryi sp2 cells and anti-PIWI antibody. The endogenous DNA and RNA that bound to NgAgo were collected, as shown in Fig. 5.

Figure 5: The DNA of ChIP and RNA of RIP were detected by 1% agarose gel electrophoresis respectively. (a) ChIP: marker (100–600 bp); Lanes 1–3, purified DNA. (b) RIP: Lane 1, negative control; Lane 2, purified RNA.
No detection of meaningful NgAgo genomic binding
We performed ChIP-seq using an anti-PIWI antibody, and the resulting sequences were aligned to the N. gregoryi sp2 genome using Bowtie 2. Enriched peaks were called using MACS2. NgAgo binding peaks were called at a threshold of P < 1.0e−2 and fragment size of approximately 200 bp. Only five binding peaks were obtained, and they could not be annotated using HOMER (Tab. 3). Peak-calling was therefore considered a failure. Peaks 1 and 5 covered genes annotated as hypothetical proteins. Peak 2 overlapped the end of Natgr_0893 (PGF-CTERM archaeal protein-sorting signal). Peak 3 included Natgr_2520 (polysulfide reductase), and peak 4 covered Natgr_2963 (transposase family protein) and Natgr_2964 (hypothetical protein).
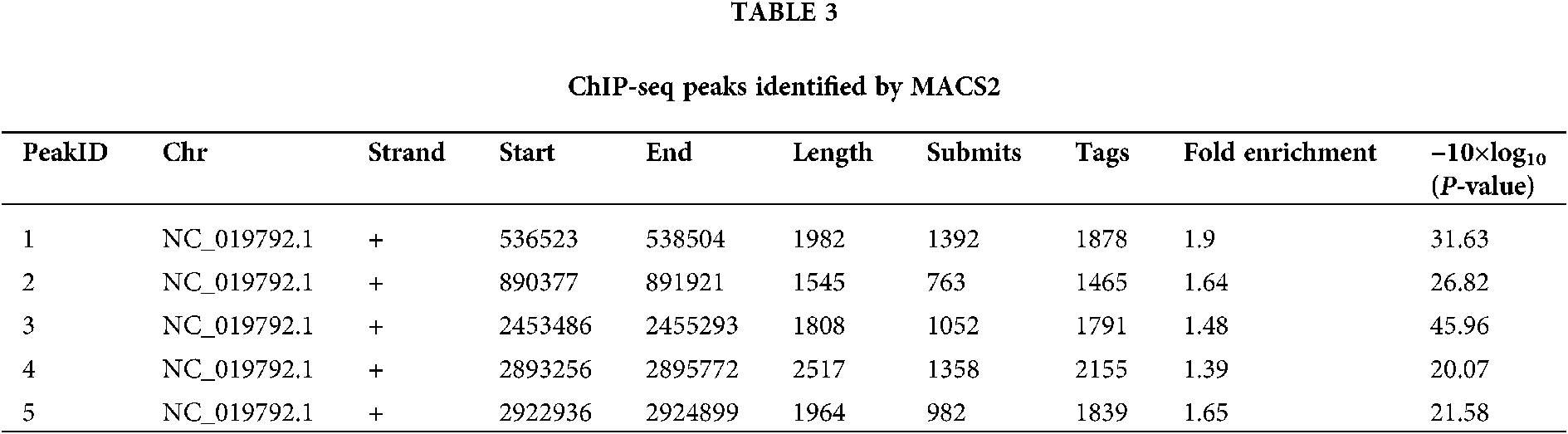
Detailed information on five peaks, including their genomic loci. Chromosome NC_019792.1 is N. gregoryi sp2. ‘Submits’ refer to the distance from the start of a peak to the summit of a peak. ‘Tags’ is the number of reads in the peak region. ‘Fold enrichment’ is the fold-change value of a peak relative to the Poisson distribution modeling background.
Endogenous transcripts bound to NgAgo
RIP-seq reads were trimmed by Trimmomatic and mapped to the N. gregoryi sp2 reference genome using TopHat. RIP-Seq gene expression was quantified as FPKM using Cufflinks. Seventeen genes were selected from a total of 3648 genes (FPKM > 1000). The expression of tRNA was the highest. The main endogenous RNAs bound to NgAgo were transcripts of tRNA, transcriptional regulators, RNA polymerase, and RNA-binding protein. These results suggest that NgAgo is involved in post-transcriptional processes in the natural state (Tab. 4).
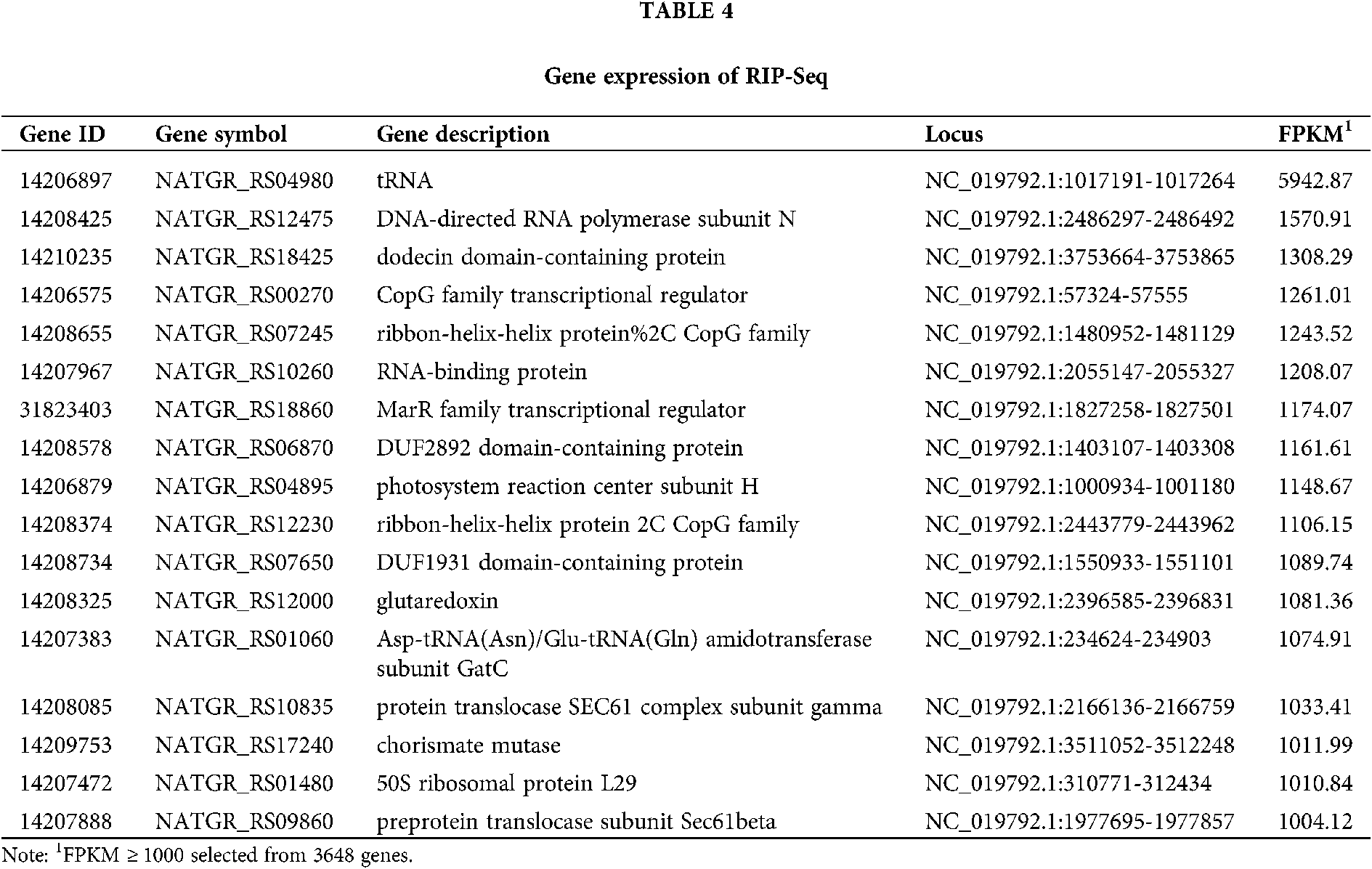
Peak detection and annotation were performed using RIPSeeker based on the .bam file obtained from the mapping procedure. RIPSeeker is a free, open-source Bioconductor/R package for RIP peak prediction based on Hidden Markov Models (HMMs). Four hundred sixty-five peaks were identified by RIPSeeker, and the peaks were classified into six categories based on their spatial positions relative to associated genes. As shown in Fig. 6, NgAgo bound mainly to transcripts inside of genes (42%), overlapping with the gene end (16%), overlapping with the gene start (20%), or upstream of genes (17%). In total, 79% of peaks were found all or in part in genic regions. In addition, coding regions accounted for 86.5% of the N. gregoryi sp2 genome.
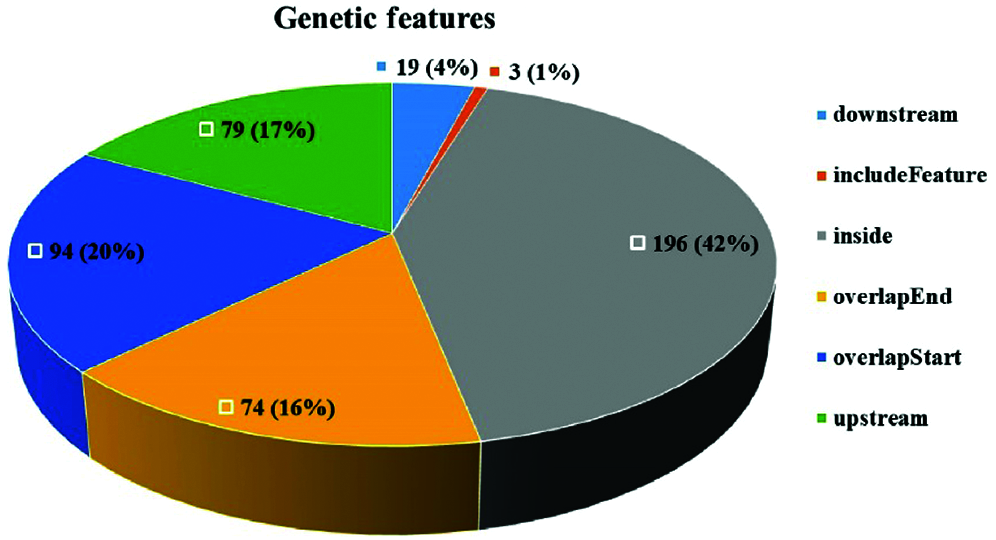
Figure 6: Peak distribution. The peaks were classified into six categories based on their spatial positions relative to associated genes.
De novo motif enrichment analysis of peaks was performed using HOMER. We searched for de novo motifs at the peaks of RIP-seq read density. There were 349 total target sequences and 20,200 total background sequences. The most enriched motif was concentrated at such peaks (Fig. 7). The top enriched motif was GGACUACAUGUU, which was most similar to Homo sapiens miR-1289 MIMAT0005879 (miRbase, Kozomara et al., 2019) with a score of 0.572. The other four top motifs were most similar to H. sapiens miR-4497, miR-4682, miR-1178, and miR-4793-3p, respectively. MicroRNAs (miRNAs) are short (20–24 nt) non-coding RNAs involved in post-transcriptional regulation of gene expression in multicellular organisms. These motifs show enrichment for G/C, A/U, and G/C residues at the seventh, ninth, and tenth positions, respectively (Fig. 8).

Figure 7: Identification of potential NgAgo binding sequences by HOMER de novo motif analysis. The top five enriched motifs out of 29 motifs (P < 1e−3). % of targets, percentage of target sequences with a given motif; % of background, percentage of background sequences with a given motif. Best match, motif matching details.
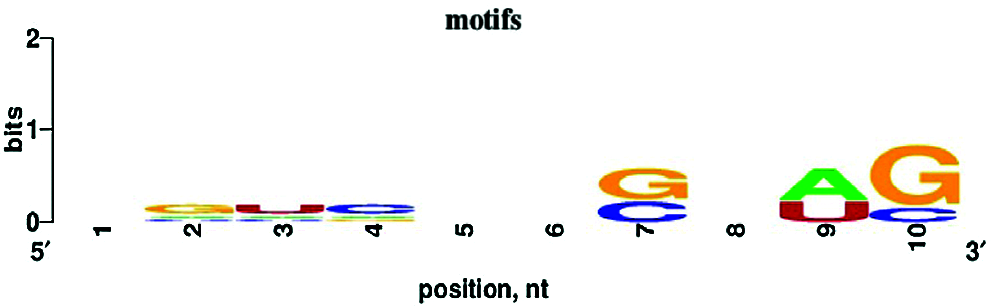
Figure 8: Nucleotide bias of the top five enriched motifs. Ten to twelve nucleotides were trimmed from the 3’ end, and the remaining 10 nt were analyzed with WebLogo.
GO term and KEGG pathway enrichment analyses were performed to annotate the functions of the peak-associated genes. The degree of enrichment was quantified by the rich factor, the P-value, and the number of genes enriched in the given term or pathway. All peak-associated genes were mapped to terms in the Gene Ontology database, the number of genes associated with each term was calculated, and the hypergeometric test was used to identify GO terms that were significantly enriched in genome peaks relative to the entire GO database background. The most enriched GO terms were related to transmembrane transport processes, including ATPase-coupled phosphate ion transmembrane transporter activity (GO:0015415), phosphate ion transmembrane transporter activity (GO:0015114), anion transport (GO:0006820), substrate-specific transmembrane transporter activity (GO:0022891), hydrolase activity, acting on acid anhydrides, catalyzing transmembrane movement of substances (GO:0016820), ATPase activity (GO:0016887), and integral component of membrane (GO:0016021) (Fig. 9). The peroxisome KEGG pathway (ko04146) was also significantly enriched (P < 0.05).
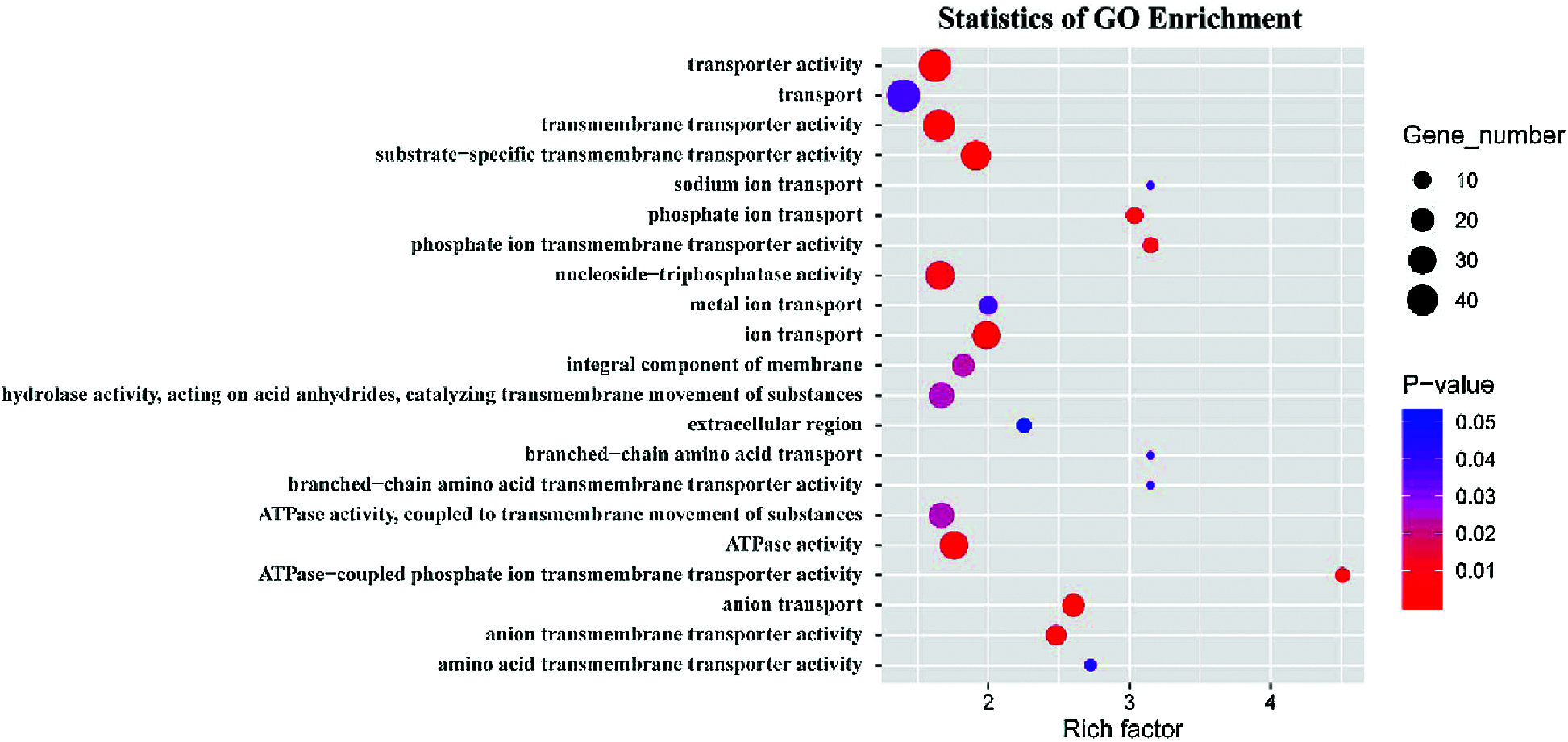
Figure 9: GO enrichment analysis showing the top 21 significantly enriched GO terms in peak-associated genes (P < 0.05).
We detected RNA but not DNA in NgAgo complexes by IP assay. Gene expression analysis of RNA-seq data indicated that NgAgo was significantly bound to ncRNA (ffs and rnpB), tRNA, and transcripts of non-histone chromosomal MC1 family protein, cold-shock protein, and twin-arginine translocation signal domain-containing protein (Tab. 2). In addition, the genes for DNA-directed RNA polymerase subunits, RNA-binding protein, and RNA-processing protein were also expressed in vivo and associated with NgAgo, as detected by IP assay. In the workflow that followed, ChIP and RIP assays were performed to specifically collect NgAgo-associated DNA and RNA, respectively. DNA was enriched in the ChIP experiment, and five enriched peaks were identified by MACS2. The peaks covered genes annotated as hypothetical protein, PGF-CTERM archaeal protein-sorting signal, polysulfide reductase, and transposase family protein. We hypothesized that such a result might be due to the use of formaldehyde cross-linking in the ChIP experiment, which fixed the NgAgo interacting protein and associated DNA. NgAgo is reported to enhance gene insertions or deletions in bacteria through the interaction of its PIWI-like domain with recombinase A (recA), thereby enhancing recA-mediated DNA strand exchange (Fu et al., 2019).
The RNA-seq expression of the IP experiment showed that NgAgo binds mainly to ncRNA (ffs and rnpB) and tRNA. The RIP-seq data indicate that NgAgo binding RNAs in N. gregoryi sp2 cells are mainly transcripts of tRNA, transcriptional regulators, RNA polymerase, and RNA-binding proteins. The tRNA-derived small RNAs (tsRNAs) are a class of novel small RNAs ubiquitously present in eukaryotes, bacteria, and archaea. The tsRNAs are involved in gene expression at the transcriptional and/or post-transcriptional levels binding to AGO or PIWI proteins in animal systems (Zhu et al., 2018). The tsRNAs are involved in the global control of small RNA silencing and inhibiting the global mRNA translation through differential Argonaute protein associations. The tsRBase was developed which is a comprehensive tsRNA repository of multiple species (Zuo et al., 2021). The tsRNA-3s are generated from the TψC loop to the 3’-end of mature tRNAs by cleavage with RNase Z or RNase P (Garcia-Silva et al., 2012; Venkatesh et al., 2016). Interestingly, the gene expression of rnpB (RNase_P_RNA) and tRNA are at a high level in the IP assay in our study. It was found that tsRNAs play a role in fundamental physiological processes such as proliferation and protein translation control (Jehn et al., 2020). The tsRNAs also play a role in pathological and physiological processes, in which gene expression is frequently dysregulated. The tsRNAs bind to Argonaute proteins and Piwi proteins like miRNAs and piRNAs sequentially (Vafaei et al., 2020). In addition, tsRNAs have been detected in archaea. The tsRNAs were generated from 51 tRNA genes encoded in halophilic archaeon Haloferax volcanii (Gebetsberger and Polacek, 2013; Heyer et al., 2012). A 26 nt-long 5’tsRNA was shown to directly bind to the small ribosomal subunit and inhibit translation by interfering with peptidyl transferase activity in H. volcanii in vitro and in vivo (Gebetsberger et al., 2012; Raina and Ibba, 2014). Here, tRNAs associated with NgAgo were both highly enriched in the IP and RIP experiments. Thus, it deserves further exploration that whether the RNAs derived from tRNA can function via binding to NgAgo in vivo.
Peak annotation indicated that NgAgo prefers to bind gene transcripts and upstream regions. It prefers the RNA motifs GGACUACAUGUU and CGUCCAGGAGGA, which are most similar to H. sapiens miR-1289 and miR-4497. Microvesicle (MV) mRNA enrichment assays have indicated that the presence of both the miR-1289 binding site and the core “CTGCC” region of a zipcode-like 25-nt sequence in the 3’ UTR of mRNAs promotes their targeting to MVs, which are considered one of the essential intercellular communication tools (Bolukbasi et al., 2012). The expression of miRNA-4497 promotes cell apoptosis (Chen et al., 2019; Tang et al., 2015; Yang et al., 2020). The complementary sequences of the top five enriched motifs were aligned to RNA sequences in the Ray2013_rbp_All_Species database using the Tomtom Motif Comparison Tool (Gupta et al., 2007). They were matched to RNCMPT00212 (P = 4.11e−3), RNCMPT00066 (P = 2.29e−4), RNCMPT00282 (P = 1.56e−2), RNCMPT00150 (P = 3.54e−3), and RNCMPT00140 (P = 5.32e−3). They were eluted from RNA-binding proteins of eukaryotes. GO enrichment showed that the peak-associated genes were enriched in transmembrane transport processes. In conclusion, we propose that endogenous nucleic acid targets of NgAgo are RNAs rather than DNAs. It is suggested that NgAgo is involved in post-transcriptional regulation processes in vivo that bear resemblance to the RNAi process of eukaryotes. However, DNA may also bind to NgAgo indirectly through other proteins such as homologous recombinase recA that interact with NgAgo.
Availability of Data and Materials: The datasets generated during the current study are available in the Sequence Read Archive (SRA) repository under the accession number PRJNA720376 (BioProject) and GenBank under the accession number PKKI00000000.
Author Contribution: Study conception and design: Lixu Jiang, Jian Huang; data collection: Lixu Jiang, Chunchao Pu, Zixin Wang; analysis and interpretation of results: Lixu Jiang, Bifang He; draft manuscript preparation: Lixu Jiang, Lin Ning. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: Not applicable.
Funding Statement: The authors are grateful to the anonymous reviewers for their valuable suggestions and comments, which have led to the improvement of this paper. This work was supported by the National Natural Science Foundation of China [Grant Nos. 61571095, 62071099, and 61901130] and the China Postdoctoral Science Foundation Grant [Grant No. 2019M653369].
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Bolger AM, Lohse M, Usadel B (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [Google Scholar]
Bolukbasi MF, Mizrak A, Ozdener GB, Madlener S, Strobel T et al. (2012). miR-1289 and “Zipcode”-like Sequence Enrich mRNAs in Microvesicles. Molecular Therapy-Nucleic Acids 1: e10. [Google Scholar]
Cyranoski D (2016). Replications, ridicule and a recluse: The controversy over NgAgo gene-editing intensifies. Nature 536: 136–137. [Google Scholar]
Chen X, Zhang L, Tang S (2019). MicroRNA-4497 functions as a tumor suppressor in laryngeal squamous cell carcinoma via negatively modulation the GBX2. Auris Nasus Larynx 46: 106–113. [Google Scholar]
Flores JK, Ataide SF (2018). Structural changes of RNA in complex with proteins in the SRP. Frontiers in Molecular Biosciences 5: 7. [Google Scholar]
Fu L, Xie C, Jin Z, Tu Z, Han L et al. (2019). The prokaryotic Argonaute proteins enhance homology sequence-directed recombination in bacteria. Nucleic Acids Research 47: 3568–3579. [Google Scholar]
Fukuda S, Yan S, Komi Y, Sun M, Gabizon R, Bustamante C (2020). The biogenesis of SRP RNA is modulated by an RNA folding intermediate attained during transcription. Molecular Cell 77: 241–250 e248. [Google Scholar]
Gagliardi M, Matarazzo MR (2016). RIP: RNA Immunoprecipitation. Methods in Molecular Biology 1480: 73–86. [Google Scholar]
Gao F, Shen XZ, Jiang F, Wu Y, Han C (2016). DNA-guided genome editing using the Natronobacterium gregoryi Argonaute. Nature Biotechnology 34: 768–773. [Google Scholar]
Garcia-Silva MR, Cabrera-Cabrera F, Guida MC, Cayota A (2012). Hints of tRNA-derived small RNAs role in RNA silencing mechanisms. Genes 3: 603–614. [Google Scholar]
Gebetsberger J, Polacek N (2013). Slicing tRNAs to boost functional ncRNA diversity. RNA Biology 10: 1798–1806. [Google Scholar]
Gebetsberger J, Zywicki M, Kunzi A, Polacek N (2012). tRNA-derived fragments target the ribosome and function as regulatory non-coding RNA in Haloferax volcanii. Archaea-an International Microbiological Journal 2012: 260909. [Google Scholar]
Geer LY, Domrachev M, Lipman DJ, Bryant SH (2002). CDART: Protein homology by domain architecture. Genome Research 12: 1619–1623. [Google Scholar]
Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS (2007). Quantifying similarity between motifs. Genome Biology 8: R24. [Google Scholar]
Hegge JW, Swarts DC, Chandradoss SD, Cui TJ, Kneppers J et al. (2019). DNA-guided DNA cleavage at moderate temperatures by Clostridium butyricum Argonaute. Nucleic Acids Research 47: 5809–5821. [Google Scholar]
Hegge JW, Swarts DC, van der Oost J (2018). Prokaryotic Argonaute proteins: Novel genome-editing tools? Nature Reviews Microbiology 16: 5–11. [Google Scholar]
Heinz S, Benner C, Spann N, Bertolino E, Lin YC et al. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell 38: 576–589. [Google Scholar]
Heyer R, Dorr M, Jellen-Ritter A, Spath B, Babski J et al. (2012). High throughput sequencing reveals a plethora of small RNAs including tRNA derived fragments in Haloferax volcanii. RNA Biology 9: 1011–1018. [Google Scholar]
Hur JK, Olovnikov I, Aravin AA (2014). Prokaryotic Argonautes defend genomes against invasive DNA. Trends in Biochemical Sciences 39: 257–259. [Google Scholar]
Javidi-Parsijani P, Niu G, Davis M, Lu P, Atala A, Lu B (2017). No evidence of genome editing activity from Natronobacterium gregoryi Argonaute (NgAgo) in human cells. PLoS One 12: e0177444. [Google Scholar]
Jehn J, Treml J, Wulsch S, Ottum B, Erb V et al. (2020). 5’ tRNA halves are highly expressed in the primate hippocampus and might sequence-specifically regulate gene expression. RNA 26: 694–707. [Google Scholar]
Jiang L, Xu H, Yun Z, Yin J, Kang J et al. (2019). Whole-genome shotgun sequence of Natronobacterium gregoryi SP2. International Conferenceon Intelligent Computing, pp. 383–393. Cham: Springer. [Google Scholar]
Jiang L, Yu M, Zhou Y, Tang Z, Li N et al. (2020). AGONOTES: A Robot Annotator for Argonaute Proteins. Interdisciplinary Sciences: Computational Life Sciences 12: 109–116. [Google Scholar]
Kaya E, Doxzen KW, Knoll KR, Wilson RC, Strutt SC et al. (2016). A bacterial Argonaute with noncanonical guide RNA specificity. Proceedings of the National Academy of Sciences of the United States of America 113: 4057–4062. [Google Scholar]
Kirsebom LA (2002). RNase P RNA-mediated catalysis. Biochemical Society Transactions 30: 1153–1158. [Google Scholar]
Kirsebom LA (2007). RNase P RNA mediated cleavage: substrate recognition and catalysis. Biochimie 89: 1183–1194. [Google Scholar]
Kozomara A, Birgaoanu M, Griffiths-Jones S (2019). miRBase: From microRNA sequences to function. Nucleic Acids Research 47: D155–D162. [Google Scholar]
Kuzmenko A, Yudin D, Ryazansky S, Kulbachinskiy A, Aravin AA (2019). Programmable DNA cleavage by Ago nucleases from mesophilic bacteria Clostridium butyricum and Limnothrix rosea. Nucleic Acids Research 47: 5822–5836. [Google Scholar]
Langmead B, Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9: 357–359. [Google Scholar]
Lee SH, Turchiano G, Ata H, Nowsheen S, Romito M et al. (2016). Failure to detect DNA-guided genome editing using Natronobacterium gregoryi Argonaute. Nature Biotechnology 35: 17–18. [Google Scholar]
Lei J, Sheng G, Cheung PP, Wang S, Li Y et al. (2019). Two symmetric arginine residues play distinct roles in Thermus thermophilus Argonaute DNA guide strand-mediated DNA target cleavage. Proceedings of the National Academy of Sciences of the United States of America 116: 845–853. [Google Scholar]
Li Y, Zhao DY, Greenblatt JF, Zhang Z (2013). RIPSeeker: A statistical package for identifying protein-associated transcripts from RIP-seq experiments. Nucleic Acids Research 41: e94. [Google Scholar]
Lisitskaya L, Aravin AA, Kulbachinskiy A (2018). DNA interference and beyond: Structure and functions of prokaryotic Argonaute proteins. Nature Communications 9: 5165. [Google Scholar]
Ma JB, Yuan YR, Meister G, Pei Y, Tuschl T, Patel DJ (2005). Structural basis for 5’-end-specific recognition of guide RNA by the A. fulgidus Piwi protein. Nature 434: 666–670. [Google Scholar]
Niaz S (2018). The AGO proteins: An overview. Biological Chemistry 399: 525–547. [Google Scholar]
O’geen H, Ren C, Coggins NB, Bates SL, Segal DJ (2018). Unexpected binding behaviors of bacterial Argonautes in human cells cast doubts on their use as targetable gene regulators. PLoS One 13: e0193818. [Google Scholar]
Olina AV, Kulbachinskiy AV, Aravin AA, Esyunina DM (2018). Argonaute proteins and mechanisms of RNA interference in eukaryotes and prokaryotes. Biochemistry 83: 483–497. [Google Scholar]
Olovnikov I, Chan K, Sachidanandam R, Newman DK, Aravin AA (2013). Bacterial argonaute samples the transcriptome to identify foreign DNA. Molecular Cell 51: 594–605. [Google Scholar]
Parker JS, Roe SM, Barford D (2005). Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature 434: 663–666. [Google Scholar]
Qi J, Dong Z, Shi Y, Wang X, Qin Y et al. (2016). NgAgo-based fabp11a gene knockdown causes eye developmental defects in zebrafish. Cell Research 26: 1349–1352. [Google Scholar]
Raina M, Ibba M (2014). tRNAs as regulators of biological processes. Frontiers in Genetics 5: 171. [Google Scholar]
Ryazansky S, Kulbachinskiy A, Aravin AA (2018). The expanded universe of prokaryotic Argonaute proteins. mBio 9: e01935–18. [Google Scholar]
Sunghyeok YEA (2017). DNA-dependent RNA cleavage by the Natronobacterium gregoryi Argonaute. http://dx.doi.org/10.1101/101923:. [Google Scholar]
Swarts DC, Jore MM, Westra ER, Zhu Y, Janssen JH et al. (2014). DNA-guided DNA interference by a prokaryotic Argonaute. Nature 507: 258–261. [Google Scholar]
Swarts DC, Koehorst JJ, Westra ER, Schaap PJ, van der Oost J (2015). Effects of Argonaute on gene expression in Thermus thermophilus. PLoS One 10: e0124880. [Google Scholar]
Swarts DC, Szczepaniak M, Sheng G, Chandradoss SD, Zhu Y et al. (2017). Autonomous generation and loading of DNA guides by bacterial Argonaute. Molecular Cell 65: 985–998 e986. [Google Scholar]
Tang L, Gao C, Gao L, Cui Y, Sha J, Liu J (2015). Expression of miRNA-4497 in human chorionic villi from early recurrent miscarriage and the influence on apoptosis. Zhonghua Yi Xue Za Zhi 95: 3737–3740. [Google Scholar]
Trapnell C, Pachter L, Salzberg SL (2009). TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [Google Scholar]
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 28: 511–515. [Google Scholar]
Vafaei S, Fattahi F, Sahlolbei M, Kiani J, Yazdanpanah A, Madjd Z (2020). Dynamic signature of tRNA-derived small RNAs in cancer pathogenesis as a promising valuable approach. Critical Reviews in Eukaryotic Gene Expression 30: 391–410. [Google Scholar]
Venkatesh T, Suresh PS, Tsutsumi R (2016). tRFs: miRNAs in disguise. Gene 579: 133–138. [Google Scholar]
Wu Z, Tan S, Xu L, Gao L, Zhu H et al. (2017). NgAgo-gDNA system efficiently suppresses hepatitis B virus replication through accelerating decay of pregenomic RNA. Antiviral Research 145: 20–23. [Google Scholar]
Yang L, Hu Z, Jin Y, Huang N, Xu S (2020). MiR-4497 mediates oxidative stress and inflammatory injury in keratinocytes induced by ultraviolet B radiation through regulating NF-κB expression, Giornale Italiano di Dermatologia e Venereologia. DOI 10.23736/S0392-0488.20.06825-X [Google Scholar] [CrossRef]
Yuan YR, Pei Y, Ma JB, Kuryavyi V, Zhadina M et al. (2005). Crystal structure of A. aeolicus argonaute, a site-specific DNA-guided endoribonuclease, provides insights into RISC-mediated mRNA cleavage. Molecular Cell 19: 405–419. [Google Scholar]
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biology 9: R137. [Google Scholar]
Zhu L, Jiang H, Sheong FK, Cui X, Gao X et al. (2016). A flexible domain-domain hinge promotes an induced-fit dominant mechanism for the loading of guide-DNA into Argonaute protein in Thermus thermophilus. Journal of Physical Chemistry B 120: 2709–2720. [Google Scholar]
Zhu L, Ow DW, Dong Z (2018). Transfer RNA-derived small RNAs in plants. Science China Life Sciences 61: 155–161. [Google Scholar]
Zuo Y, Zhu L, Guo Z, Liu W, Zhang J et al. (2021). tsRBase: A comprehensive database for expression and function of tsRNAs in multiple species. Nucleic Acids Research 49: D1038–D1045. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |