DOI:10.32604/biocell.2021.014844
| Biocell DOI:10.32604/biocell.2021.014844 | |
| Article |
KRT4 suppresses oral squamous cell carcinoma development by reducing ATG4B-mediated autophagy
Guangdong Provincial Key Laboratory of Stomatology, Guanghua School of Stomatology, Hospital of Stomatology, Sun Yat-Sen University, Guangzhou, 510055, China
*Address correspondence to: Bin Cheng, chengbin@mail.sysu.edu.cn
Received: 03 November 2020; Accepted: 12 January 2021
Abstract: Head and neck squamous cell carcinoma is the sixth most common tumor worldwide, and half of head and neck squamous cell carcinoma patients are with oral squamous cell carcinoma (OSCC). 300,000 new cases of OSCC were reported annually. Even with multi-modality treatment, the prognosis of OSCC remains unsatisfactory. Thus, it is urgent to discover novel therapeutic targets for OSCC. Some microarray studies have revealed that Keratin 4 (KRT4) is downregulated in OSCC, whereas its role in OSCC development remains unknown. The present study revealed that KRT4 suppressed OSCC progression by inducing cell apoptosis and inhibiting cell invasion. In addition, KRT4 over-expression inhibited autophagy by blocking the interaction of autophagy-related 4B cysteine peptidase (ATG4B) and microtubule-associated protein 1A/1B light chain 3 (LC3) to regulate apoptosis and invasion of OSCC. In conclusion, KRT4 played an important role in OSCC development through regulating ATG4B-mediated autophagy and may be a novel therapeutic drug target of OSCC.
Keywords: Oral squamous cell carcinoma; KRT4; Cancer-suppressive role; ATG4B; Autophagy
Head and neck squamous cell carcinoma is the sixth most prevalent tumor worldwide, and half of those occur in the oral cavity (Haddad and Shin, 2008; Kademani, 2007). OSCC accounts for more than 90% of all oral malignancies and 2–3% of all cancers, with more than 300,000 new cases reported annually (Choi and Myers, 2008; Ferlay et al., 2010). In China, over 14,000 deaths due to OSCC are reported each year (Zheng et al., 2015). With the development of cytobiology and molecular biology, therapies of OSCC have been significantly improved. However, more than 50% of OSCC patients still die under current therapies (Chiou et al., 2008; Lo et al., 2003), as the development of OSCC is a multi-step process, always going through stages of normal mucosa, precancerous lesions, and cancer (Argiris et al., 2008; Hashibe et al., 2009). Thus, it is urgent to elucidate novel mechanisms regulating OSCC development and search for promising biomarkers for targeted therapies of OSCC.
Technological breakthroughs, including laser capture microdissection and microarray technology, have fundamentally altered cancer research, which allows the high-throughput analysis of thousands of genes. Several studies have revealed that KRT4 was downregulated in OSCC (Lallemant et al., 2009; Toruner et al., 2004; Ye et al., 2008). KRT4 belongs to the Type II keratin, which is specifically expressed with family member KRT13 in differentiated layers of the mucosal and esophageal epithelia (Zhang et al., 2018a). Mutations of these genes would lead to White Sponge Nevus (WSN) (Zhang et al., 2018a). A previous study has revealed that KRT4 serves as a tumor marker in esophageal squamous cell carcinoma (Takikita et al., 2011). However, the role of KRT4 in OSCC remains unknown.
Through affinity capture-MS detection, KRT4 is found to interact with ATG4B (Behrends et al., 2010). ATG4B is required for autophagy and serves as a cysteine protease for hydrolyzing LC3I to LC3II or for the deconjugation of LC3II (Nakatogawa et al., 2007; Pengo et al., 2017). Autophagy mediates bulk degradation involved in the constitutive clearance of intracellular components, including damaged cellular organelles and protein aggregations, and plays an important role in maintaining cellular homeostasis (Mizushima et al., 2008; Weidberg et al., 2011). Dysfunction of autophagy leads to many diseases, such as cancer (Levine and Kroemer, 2019). In different contexts, autophagy plays neutral, suppressive, or promoting roles in cancer. For example, autophagy could maintain tumor growth in a nutrient-limited micro-environment (Amaravadi et al., 2016). Numerous studies have indicated that elevated expression of ATG4B enhances tumor growth, migration, and invasion by inducing autophagy in many types of cancer (Akin et al., 2014; Levine, 2007; Tran et al., 2013). To date, the role of ATG4B in OSCC is little known.
The aim of this study was to investigate the role of KRT4 in OSCC development. We found that KRT4 was downregulated in OSCC cells. Overexpression of KRT4 significantly induced OSCC cell apoptosis and suppressed cell invasion of OSCC cells. Moreover, KRT4 regulated apoptosis and invasion in OSCC cells by suppressing autophagy via blocking the interaction of ATG4B and LC3. These results revealed that KRT4 suppressed OSCC development through regulating autophagy.
Normal oral keratinocytes (NOK) and OSCC cells (HN6) obtained from Cell Bank at the Chinese Academy of Sciences (Shanghai, China) were cultured with DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin in a humidified atmosphere of 5% CO2 at 37°C.
Cells were transfected with the KRT4 expression vector, ATG4B expression vector, or ATG4B siRNAs using Lipofectamine 2000 (Invitrogen, Carlsbad, USA), according to the manufacturer’s instruction. After 48 h, cells were collected for further experiments.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cells with TRIzol reagent (Invitrogen). Next, cDNA was synthesized using SYBR ExScript RT-PCR kit (Toyobo, Japan). PCR primers for KRT4 (forward: 5’-CTCCAGCAAAAACCTTGAGC-3’; reverse: 5’-AAGTCATTCTCGGCTGCTGT-3’) and ATG4B (forward: 5’-ACTGATGGCTGCTTCATCCC-3’; reverse: 5’-AGAATCTAGGGACAGGTTCAGGA-3’) were synthesized. GAPDH (forward: 5’-GCACCGTCAAGGCTGAGAAC-3’; reverse: 5’-ATGGTGGTGAAGACGCCAGT-3’) was used as a reference to normalize amounts of target RNAs. qRT-PCR was performed by the ABI7300 Real-Time PCR system (Applied Biosystems, Shanghai, China) using SYBR green fluorophore. Threshold cycle (Ct) data were collected, and mRNA fold change (FC) relative to GAPDH was calculated by the comparative Ct method of 2−ΔΔCt.
Equal amounts of protein from different groups were resolved by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto Immobilon-P membranes (Millipore, Shanghai, China). The membranes were blocked with 5% nonfat milk at room temperature (RT) for 1 h and subsequently incubated with the following primary antibodies in 1% nonfat milk at 4°C overnight: Rabbit anti-KRT4 (1:500), mouse anti-ATG4B (1:400), rabbit anti-LC3 (1:500). All primary antibodies were obtained from Abcam (Boston, USA). Following primary antibody incubation, the membranes were washed for 45 min with Tris-buffered saline containing 0.1% Tween20 (TBS-T) and then exposed to goat anti-rabbit or mouse secondary antibody. The signals of proteins were detected by the SuperSignal West Femto Maximum Sensitivity Substrate kit (Thermo Scientific, Rockford, USA).
Cell Counting Kit-8 (CCK8) assay
The cell viability was detected using CCK8 assay following the manufacturer’s protocol. Briefly, cells were seeded in a 96-well culture plate (5 × 103 cells/well) for 24 h. Next, 10 μL of CCK-8 reagent (Beyotime, Shanghai, China) was added into each well. Subsequently, cells were incubated at 37°C for 2 h, followed by measuring the absorbance (optical density, OD) at 450 nm on an enzyme immunoassay analyzer (Bio-Rad).
Flow cytometric apoptosis assay
1 × 106 cells from each group plated in 24-well plates were collected and washed twice using incubation buffer (10 mmol/L HEPES/NaOH, pH 7.4, 140 mmol/L NaCl, 5 mmol/L CaCl2). Subsequently, cells were resuspended into 100 μL phosphate-buffered saline (PBS) containing 1.5 μg/mL Annexin V (FACSARIA, BD Biosciences, San Jose, USA) and moderate Propidium iodide (PI) (Thermo Scientific Rockford, USA) and incubated at RT for 10–15 min in the dark. After washing by PBS three times, cells were resuspended by incubation buffer and analyzed by flow cytometry (BD Biosciences).
Cell migration was tested by the scratch wound-healing assay. 5 × 105 cells were grown in 35 mm plates and followed by transfection of expression vectors for 2 h. Then a scratch wound was created in two perpendicular directions by scraping the cell monolayer using a pipette tip, and the culture medium was replenished immediately. After 48 h, phase-contrast images of the cells were captured. Then wound width was calculated as the average distance between the sides of the scratches measured by ImageJ (https://imagej.nih.gov/ij/). Subsequently, the migration rate was identified through decreased wound width dividing the duration of migration.
Cells were suspended in 200 μL DMEM medium without FBS and seeded in the upper compartment Transwell inserts (Millipore) coated with Matrigel (Corning, Shanghai, China). Meanwhile, the lower chamber was filled with DMEM medium supplemented with 10% FBS. After 24 h incubation at 37°C, noninvasive cells were cleaned by cotton swabs, and the invasive cells in the lower chamber were fixed with 4% paraformaldehyde followed by the staining of 0.5% crystal violet for 10 min at RT. Next, invasive stained cells were photographed and counted by a light microscope (Leica, Heidelberg, Germany).
Co-Immunoprecipitation (Co-IP)
Co-IP was performed as previously described (Tang and Takahashi, 2018). 300 mg of protein from cells, protease inhibitor cocktail (Sigma-Aldrich, Shanghai, China), lysis buffer (Cell Signaling Technology, Beverly, USA), Protein A/G Magnetic bead slurry (Thermos Fisher Scientific Rockford, USA), 1 μg mouse anti-ATG4B antibody (Abcam), 1 μg rabbit anti-LC3 antibody (Abcam) and mouse IgG (Cell Signaling Technology) were used.
Cells were fixed in 4% paraformaldehyde for 20 min at RT, followed by PBS washing three times. Then cells were blocked by 10% donkey serum albumin in PBST (0.1% Triton X-100 in PBS) for 1 h at RT. The primary antibody LC3 antibody (1:200) was diluted in PBS supplementary with 1% donkey serum and incubated with cells at 4°C overnight. Next, cells were washed by PBST five times and incubated with secondary antibody in the dark at RT. Finally, cells were counterstained with DAPI and mounted with an aqueous mounting medium (Sigma-Aldrich). Images were captured by an inverted microscope (Leica).
All data in this study were representative of three repeated experiments. The quantitative data were present as means ± standard deviation (SD). Statistical differences were analyzed by the unpaired Student’s t-test between two groups or one-way ANOVA followed by Student-Newman-Keuls post hoc test between multiple groups. Moreover, all statistical analyses were carried out via the SPSS 20.0 software (SPSS Inc., Chicago, USA). The value of p < 0.05 indicated statistical significance.
KRT4 overexpression induces apoptosis in OSCC cells
Compared to NOK cells, the KRT4 mRNA and protein levels were significantly downregulated in HN6 cells (Figs. 1A and 1B). Moreover, overexpression of KRT4 did not regulate HN6 cell proliferation, whereas suppressed NOK cell viability, detected by CCK8 assay (Fig. 1C). Furthermore, cell apoptosis was performed by flow cytometric analysis to count the rate of early and late apoptotic cells. Results showed that KRT4 overexpression increased the rate of early and late apoptotic HN6 cells from 9.6% to 18.8%, whereas KRT4 had no effect on NOK cell apoptosis (Fig. 1D). These results suggested that KRT4 enhanced apoptosis in OSCC cells.
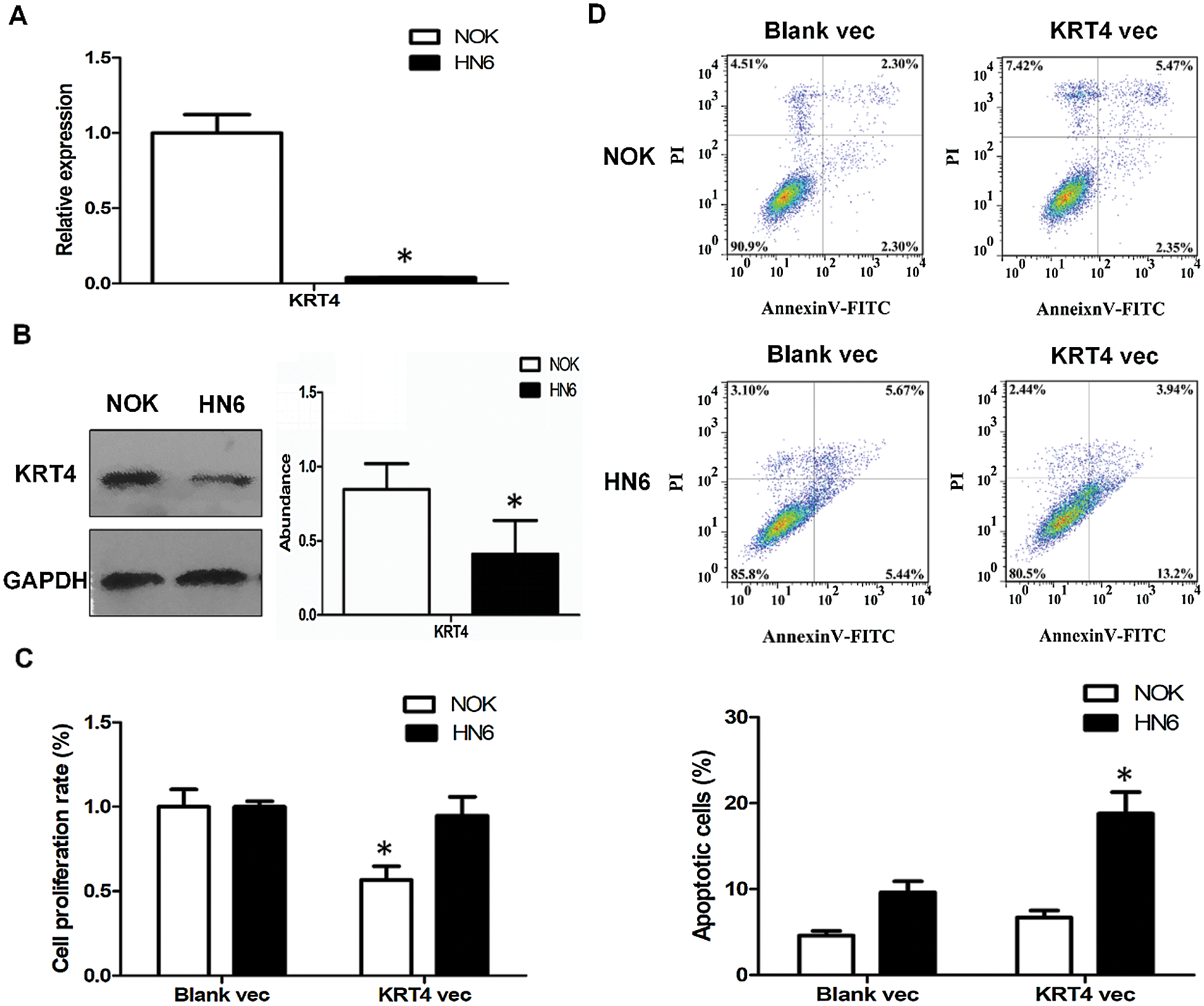
Figure 1: KRT4 overexpression triggers apoptosis of OSCC.
(A) qRT-PCR analysis of KRT4 mRNA levels in NOK and HN6 cells. (B) WB analysis of KRT4 protein levels in NOK and HN6 cells. The bar graph showed the quantification of KRT4 protein levels normalized to GAPDH. (C) CCK8 assay analysis of cell growth in NOK and HN6 cells. The bar graph showed the cell proliferation rate. (D) Flow cytometric analysis of apoptosis in NOK and HN6 cells. The bar graph showed the number of apoptotic cells. Vec, expression vector. *p < 0.05.
KRT4 overexpression suppresses invasion of OSCC cells
To identifying the role of KRT4 in migration and invasion of OSCC cells, scratch wound healing assay and Transwell assay were performed. Results indicated that overexpression of KRT4 did not affect HN6 cell migration (Fig. 2A). Furthermore, KRT4 overexpression reduced the invasive HN6 cell number from 157 to 93 (Fig. 2B). The above data suggested that KRT4 inhibited the invasion of OSCC cells.
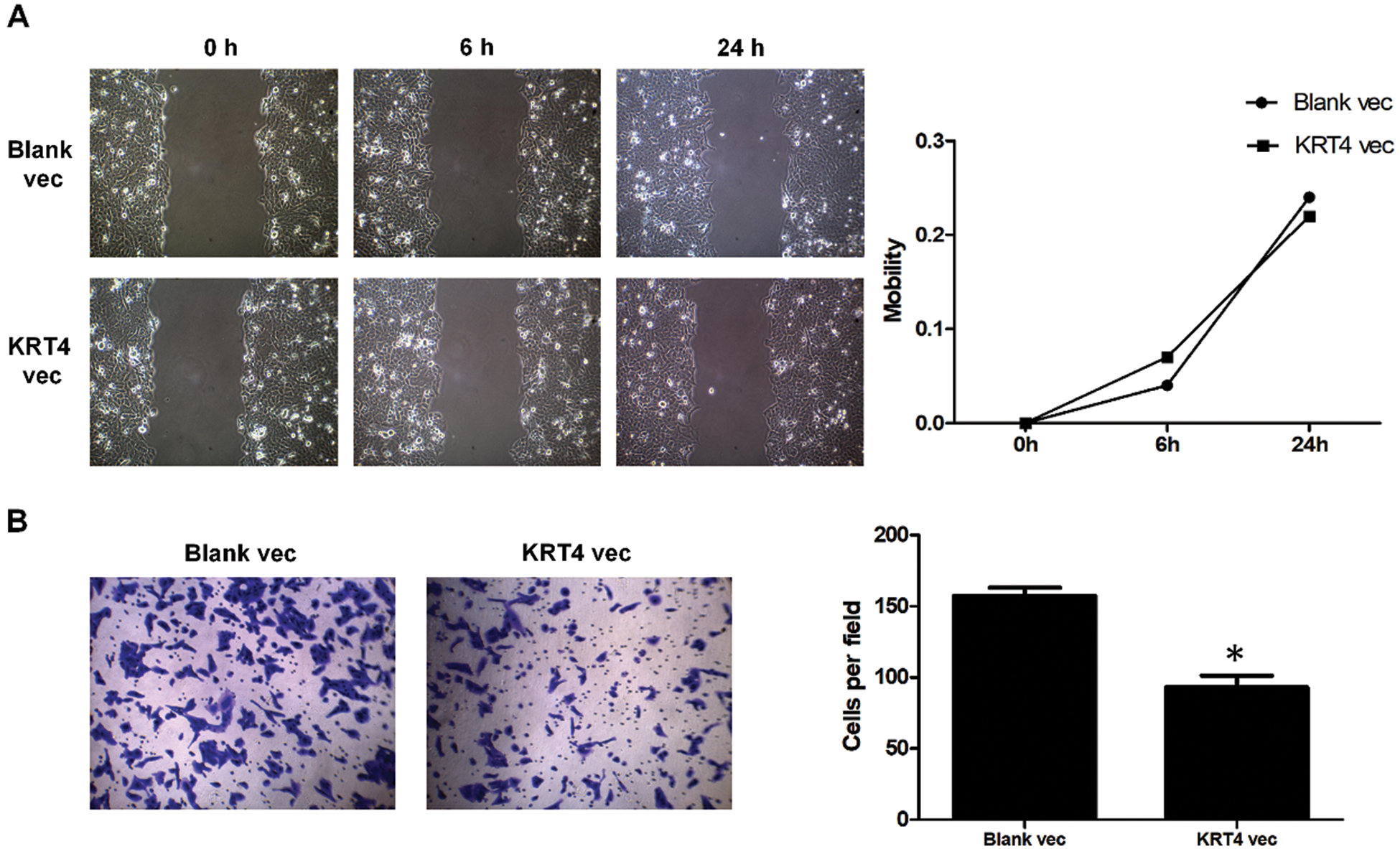
Figure 2: Overexpression of KRT4 suppresses OSCC invasion and KRT4 protein associates with ATG4B protein.
(A) Scratch wound healing assay analysis of cell migration of KRT4-overexpressed HN6 cells. The line graph showed the migration rate. (B) Transwell assay analysis of cell invasion of KRT4-overexpressed HN6 cells. The bar graph showed the number of invasive cells. cellscellsVec, expression vector. *p < 0.05.
KRT4 protein interacts with ATG4B protein in OSCC cells
The above results had demonstrated the cancer-suppressive role of KRT4 in OSCC. Next, the mechanism of how KRT4 suppressed OSCC development was explored. A previous study has demonstrated that KRT4 protein associates with ATG4B protein in 293T cells (Behrends et al., 2010). Thus, we investigated whether KRT4 protein interacted with ATG4B protein in HN6 cells by Co-IP using a KRT4 antibody. Results revealed that KRT4 protein bound the ATG4B protein in HN6 cells (Fig. 3A). In addition, the expression of ATG4B detected by qPCR and WB was not changed in HN6 cells compared to that in NOK cells (Figs. 3B and 3C). These results together suggested that KRT4 may exert a tumor-suppressive effect through ATG4B.
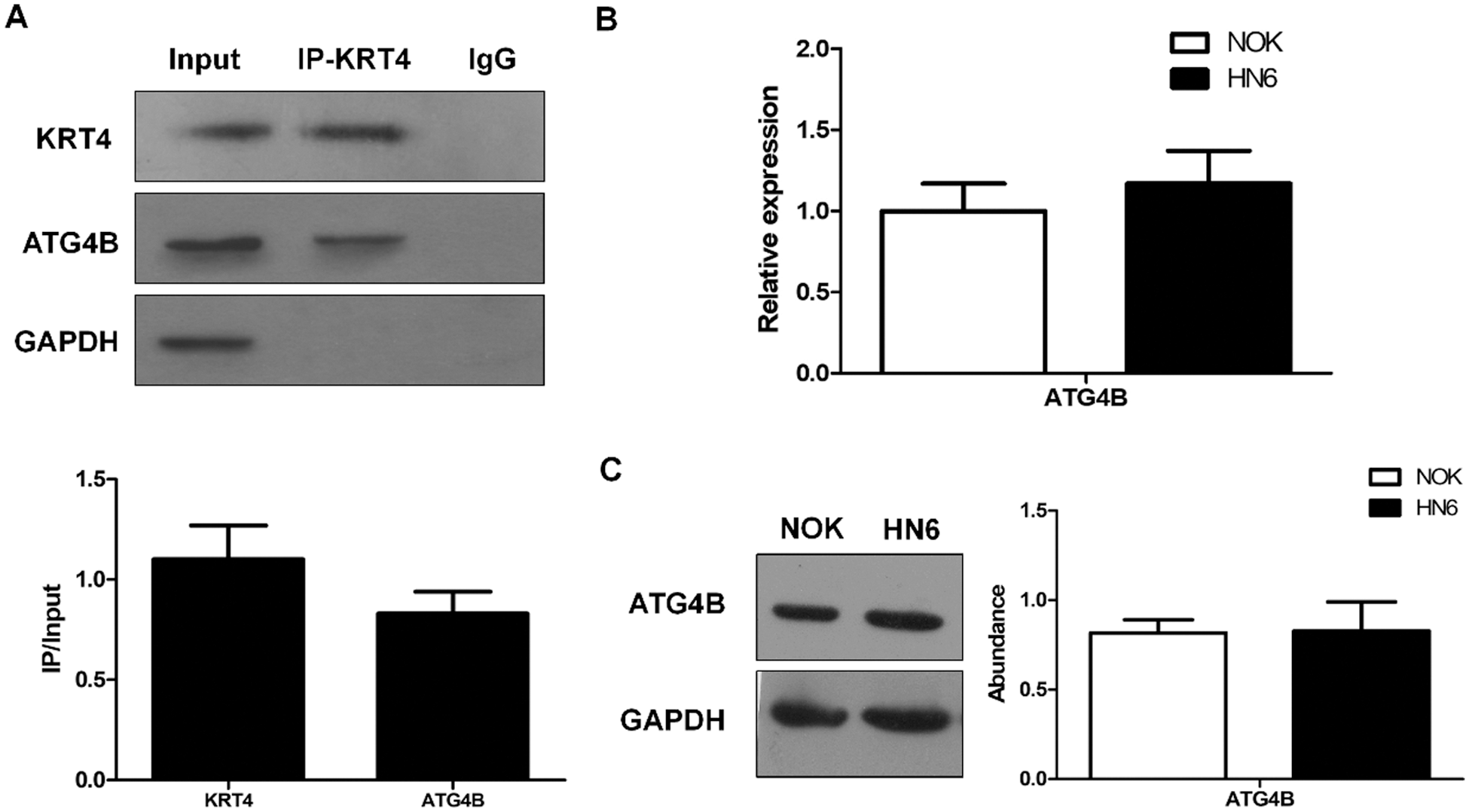
Figure 3: ATG4B silence triggers apoptosis and inhibits invasion of OSCC.
(A) Co-IP analysis of protein interaction between KRT4 and ATG4B using a KRT4 antibody. The bar graph showed the KRT4 or ATG4B protein levels in KRT4 immunoprecipitates normalized to 10% input. (B) qRT-PCR analysis of ATG4B mRNA levels in NOK and HN6 cells. (C) WB analysis of ATG4B protein levels in NOK and HN6 cells. The bar graph showed the quantification of ATG4B protein levels normalized to GAPDH. *p < 0.05.
Silence of ATG4B triggers apoptosis and suppresses invasion in OSCC cells
The role of ATG4B in OSCC remains unclear so far. To identify the effect of ATG4B on OSCC cells, siRNAs were transfected into HN6 cells to knockdown ATG4B. Among the three siRNAs, ATG4B siRNA1 was found to have the most powerful effect (Fig. 4A), which was used for subsequent experiments. Further results revealed that ATG4B silence increased the rate of early and late apoptotic HN6 cells from 4.58% to 13.95%, suggesting that ATG4B induced HN6 cell apoptosis (Fig. 4B). Moreover, ATG4B silencing slightly inhibited HN6 cell migration (Fig. 4C). Besides, the silencing of ATG4B reduced the number of invasive HN6 cells from 203 to 96, significantly suppressed HN6 cell invasion (Fig. 4D). These results suggested that ATG4B silence induced apoptosis and inhibited invasion in OSCC cells, which were consistent with those in KRT4-overexpressed HN6 cells.
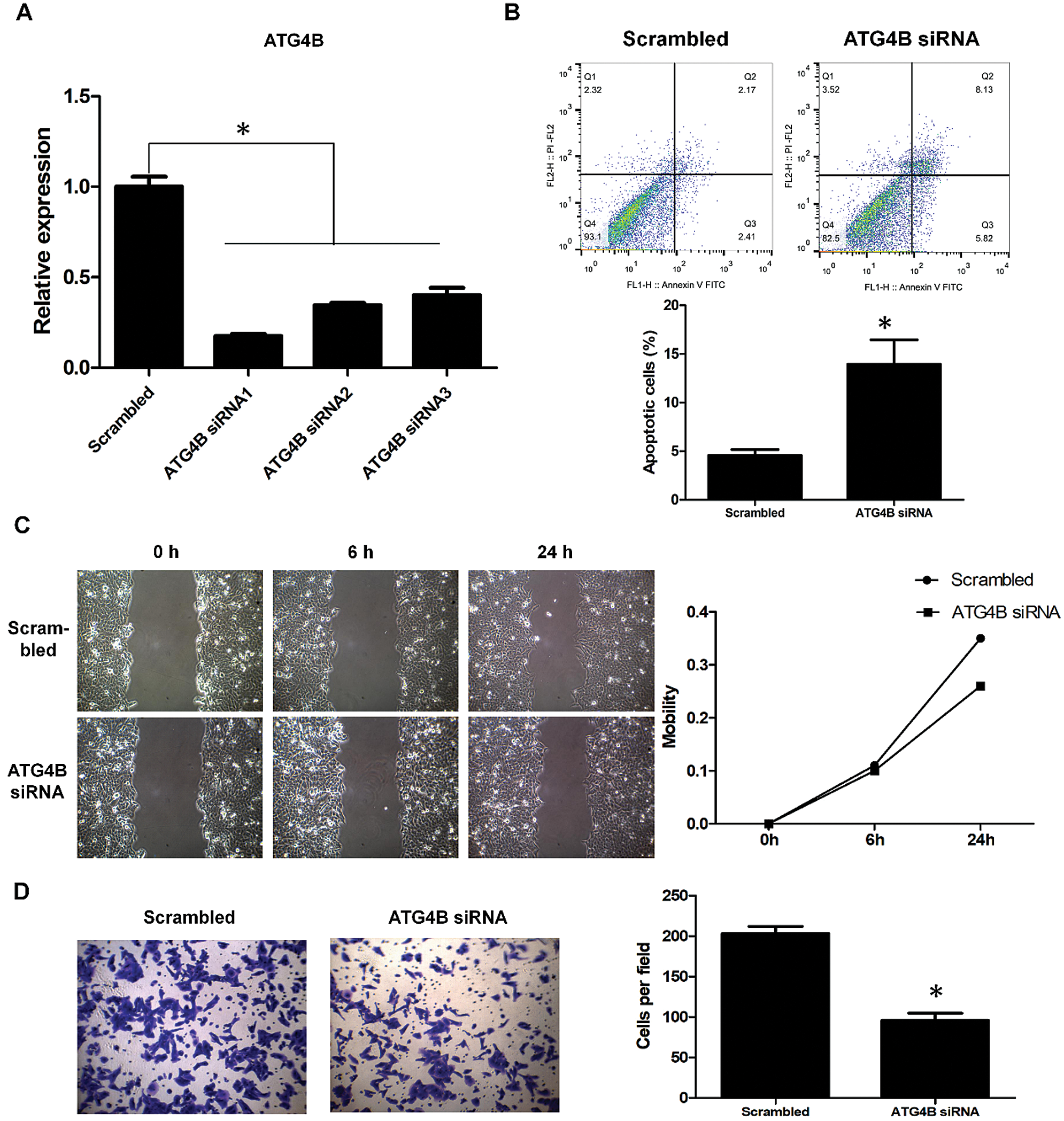
Figure 4: ATG4B silence triggers apoptosis and inhibits invasion of OSCC.
(A) qRT-PCR analysis of ATG4B mRNA levels in HN6 cells transfected with ATG4B siRNAs. (B) Flow cytometric analysis of apoptosis in HN6 cells transfected with ATG4B siRNAs. The bar graph showed the number of apoptotic cells. (C) Scratch wound healing assay analysis of cell migration in HN6 cells transfected with ATG4B siRNAs. The line graph showed the migration rate. (D) Transwell assay analysis of cell invasion in HN6 cells transfected with ATG4B siRNAs. The bar graph showed the number of invasive cells. *p < 0.05.
Furthermore, ATG4B overexpression reversed the effect of KRT4 overexpression on HN6 cell apoptosis (Fig. 5A). Transwell invasion assay also revealed that ATG4B overexpression abolished the effect of KRT4 overexpression on HN6 cell invasion (Fig. 5B). All these data indicated that KRT4 regulated OSCC development by inhibiting the function of ATG4B.
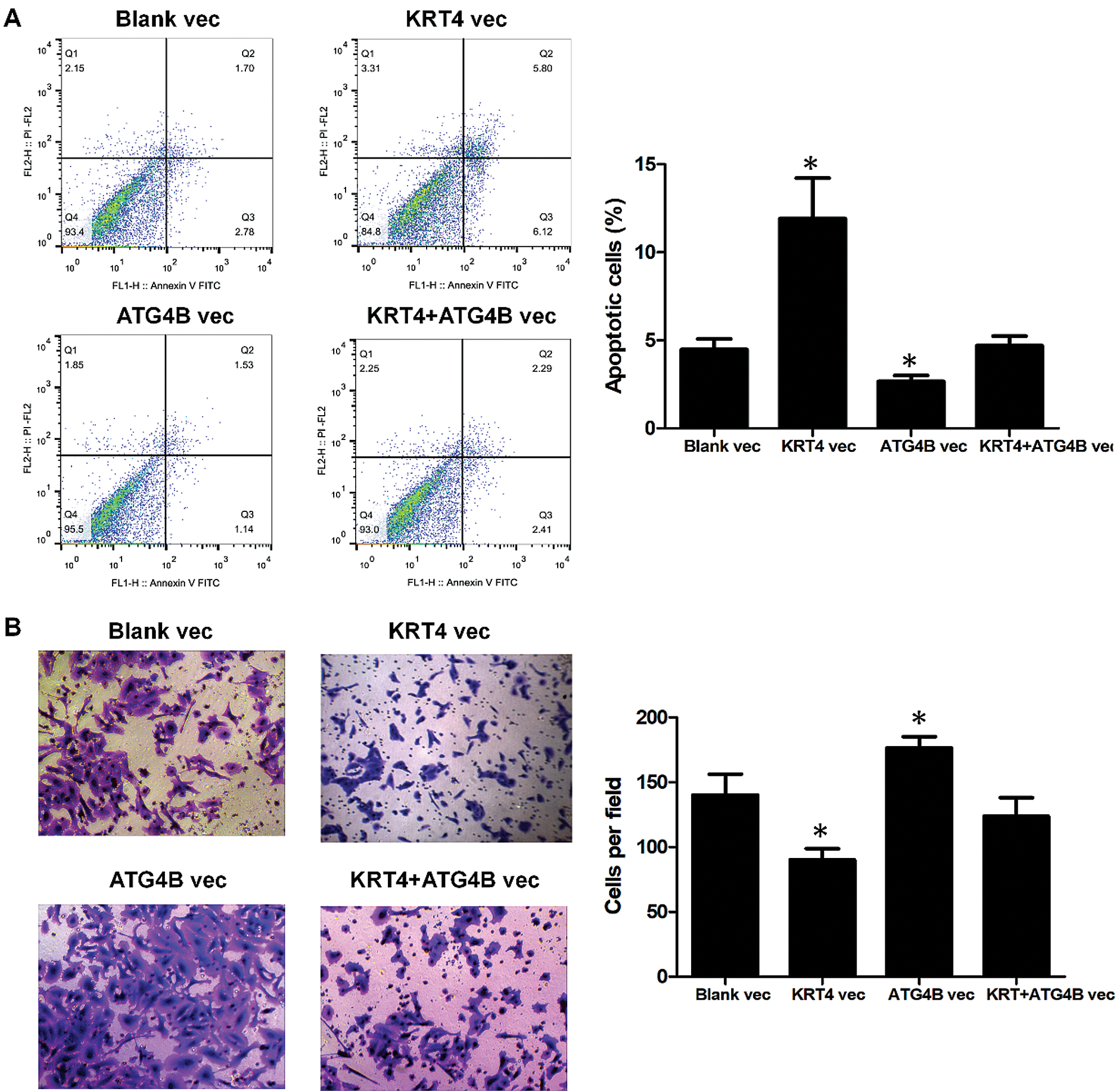
Figure 5: ATG4B overexpression reverses the effect of KRT4 overexpression.
(A) Flow cytometric analysis of apoptosis in ATG4B-overexpressed or ATG4B plus KRT4-overexpressed HN6 cells. The bar graph showed the number of apoptotic cells. (B) Transwell assay analysis of cell invasion in ATG4B-overexpressed or ATG4B plus KRT4-overexpressed HN6 cells. The bar graph showed the number of invasive cells. Vec, expression vector. *p < 0.05.
KRT4 overexpression reduces autophagy in OSCC cells
As ATG4B is required for autophagy, we detected the effect of KRT4 on autophagy of HN6 cells. The number of autophagosomes that responded to autophagy levels was detected through LC3 immunostaining kin HN6 cells. Results showed that KRT4 overexpression dramatically decreased the number of LC3 green puncta indicated autophagosome in HN6 cells (Fig. 6A). Moreover, the level of LC3 II required for autophagosome formation was also reduced in KRT4-overexpressed HN6 cells (Fig. 6B). Consistently, ATG4B silence also decreased the number of autophagosomes and reduced LC3II level (Figs. 6C and 6D). All these results suggested that KRT4 suppressed autophagy through ATG4B in OSCC cells.
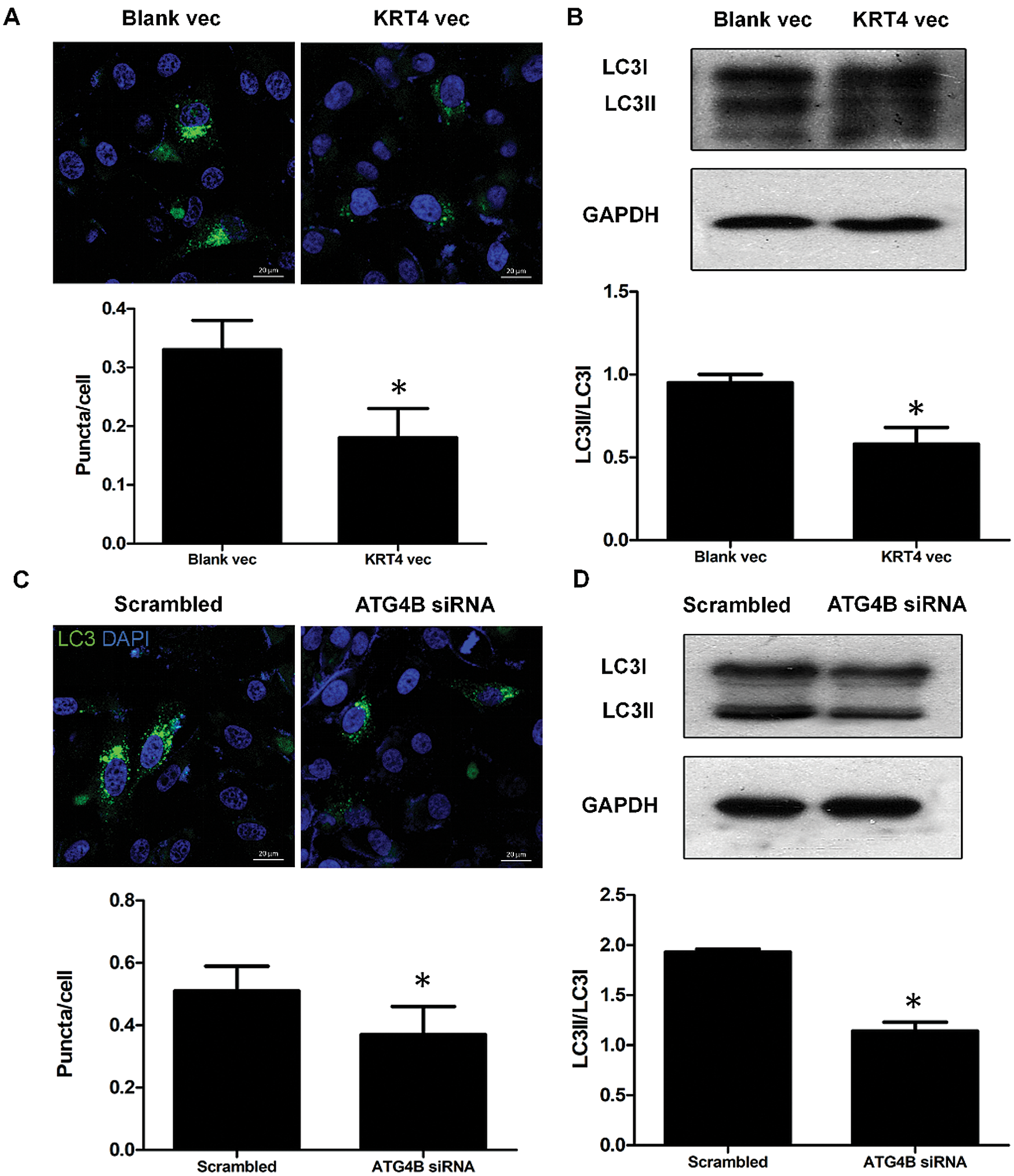
Figure 6: KRT4 overexpression decreases the autophagy level of OSCC.
(A) Immunostaining analysis of LC3 puncta indicated autophagosome in HN6 cells transfected with or without KRT4 expression vector. The bar graph showed the average number of LC3 puncta per cell. (B) WB analysis of LC3 protein levels in HN6 cells transfected with or without KRT4 expression vector. The bar graph showed the quantification of LC3II protein levels normalized to LC3I. (C) Immunostaining analysis of LC3 puncta indicated autophagosome in HN6 cells transfected with or without ATG4B siRNA. The bar graph showed the average number of LC3 puncta per cell. (D) WB analysis of LC3 protein levels in HN6 cells transfected with or without ATG4B siRNA. The bar graph showed the quantification of LC3II protein levels normalized to LC3I. *p < 0.05.
KRT4 suppresses autophagy through inhibiting the interaction of ATG4B and LC3
ATG4B facilitates autophagy through binding with LC3 to hydrolyze LC3I to LC3II or deconjugate LC3II. To further demonstrate the mechanism of how KRT4 suppressed autophagy in HN6 cells, the interaction of ATG4B and LC3 (including LC3I and LC3II) was detected by Co-IP. Compared to the control group (Group 2), KRT4 overexpression (Group 1) reduced the interaction between ATG4B and LC3 (Fig. 7), suggesting that KRT4 prohibited autophagy by disrupting the interaction between ATG4B and LC3.
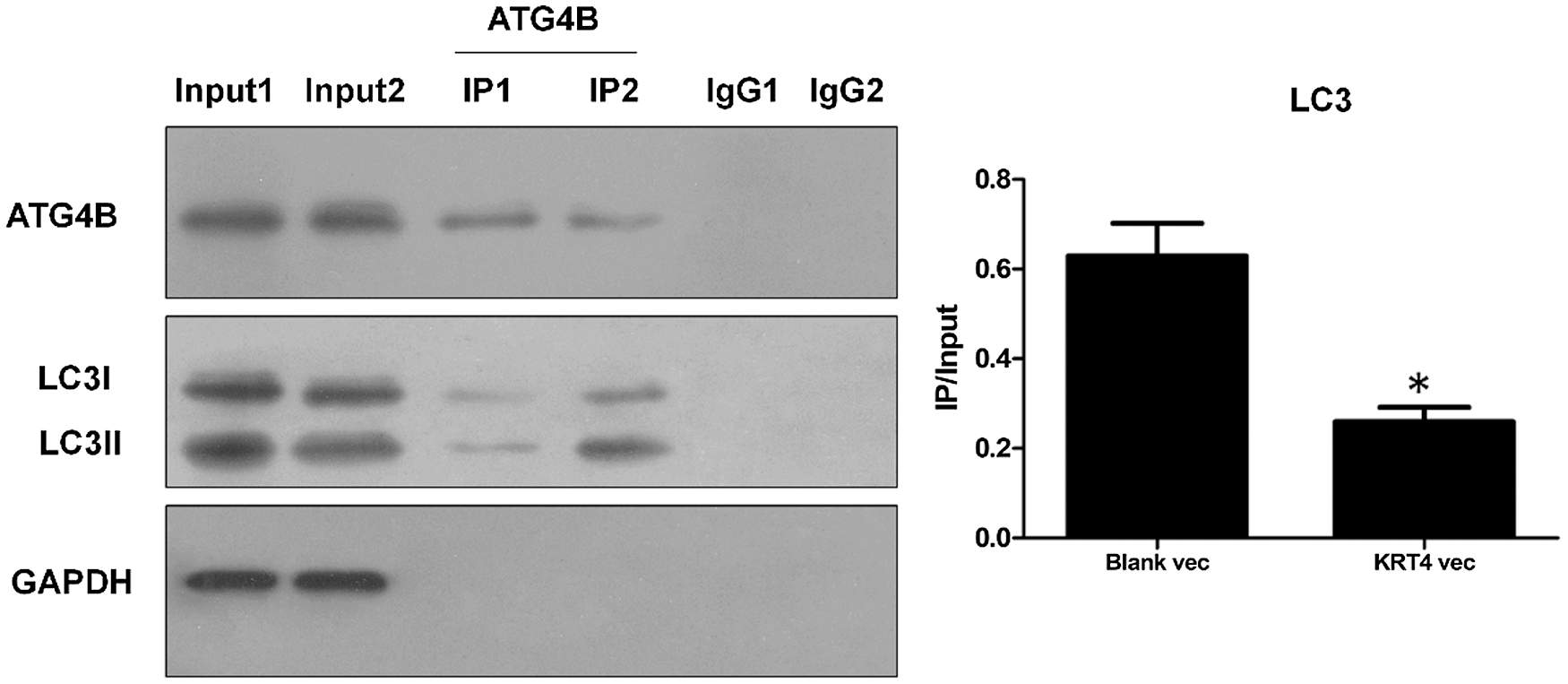
Figure 7: KRT4 reduces autophagy by blocking the interaction between ATG4B and LC3.
Co-IP analysis of protein interaction between ATG4B and LC3 using an ATG4B antibody. The bar graph showed the LC3 (both LC3I and LC3II) protein levels in ATG4B immunoprecipitates normalized to 10% input. Group 1: HN6 cells transfected with KRT4 overexpression vector, Group 2: HN6 cells transfected with the blank expression vector. *p < 0.05.
KRT4 is known as the inactivator of WSN characterized by benign oral, esophageal, and anal leukoplakia (Chao et al., 2003; Kimura et al., 2013; Zhang et al., 2018a; Zhang et al., 2009). However, the role of KRT4 in OSCC is little known, though its transcription is significantly reduced in OSCC. In the present study, we demonstrated that KRT4 played a cancer-suppressive role in OSCC through inducing cell apoptosis and suppressing cell invasion. This study further revealed that KRT4 suppressed OSCC development by inhibiting ATG4B-mediated autophagy via disrupting the interaction between ATG4B and LC3. This study revealed the cancer-suppressive role of KRT4 for the first time.
Usually, induction of apoptosis leads to the reduction of cell proliferation. However, our results indicated a dramatic induction of apoptosis in HN6 cells caused by KRT4 overexpression, while the proliferation of HN6 cells remained unaffected by KRT4 overexpression. Some previous studies have revealed a similar finding. For example, terazosin and doxazosin trigger prostate apoptosis without modifying cell proliferation in patients with benign prostatic hyperplasia (Chon et al., 1999). In addition, the apoptotic index of emphysematous lungs is significantly higher than that of the control group, whereas the proliferation index is consistent in emphysema patients and healthy people (Calabrese et al., 2005). Therefore, induction of apoptosis may not affect cell proliferation.
In different contexts, autophagy plays neutral, suppressive, or promoting roles in cancer (Amaravadi et al., 2016). Several studies have revealed that deletion of autophagy-related gene Atg5 or Atg7 in mice induces benign tumors, suggesting that autophagy suppresses cancer initiation (Akito et al., 2011; Rosenfeld et al., 2014). In addition, autophagy could promote the growth of advanced cancer, such as deletion of Atg7 reduces oncogenic Kras-driven lung tumor cell proliferation (Guo et al., 2013). Moreover, impaired autophagy caused by monoallelic loss of Becn1 suppresses Palb2-associated mammary tumorigenesis (Huo et al., 2013). Our results indicated that KRT4 suppressed OSCC development by inhibiting ATG4B-mediated autophagy. Thus, ATG4B-mediated autophagy played tumor-promoting roles in OSCC.
As mentioned above, KRT4 induced OSCC apoptosis by suppressing autophagy. Numerous studies have revealed that inhibition of autophagy enhances apoptosis. For example, inhibiting reactive oxygen species (ROS)-dependent autophagy promotes baicalein-induced apoptosis in OSCC (Li et al., 2017). Nimbolide, an inhibitor of nuclear factor kappa-B (NF-κB), also suppresses cytoprotective autophagy to activate apoptosis via the phosphoinositide-3-kinase (PI3K)/AKT serine/threonine kinase (AKT)/Glycogen synthase kinase-3β (GSK-3β) signaling pathway (Sophia et al., 2018). In addition, RANK ligand (RANKL) induces autophagy to promote resistance for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptotic cell death in OSCC (Ethiraj et al., 2019). Furthermore, epidermal growth factor receptor (EGFR) confers radio-resistance by enhancing autophagy and suppressing apoptosis through endoplasmic reticulum (ER) stress signaling PERK-eukaryotic initiation factor 2α (eIF2α)-glucose regulated protein 94 (GRP94) and endoplasmic reticulum to nucleus signaling 1 (IRE1α)- X-box binding protein 1 (XBP1)-glucose regulated protein 78 (GRP78) (Zhang et al., 2018b). Therefore, PI3K/AKT/GSK-3β, TRAIL, or ER stress signaling pathways may contribute to KRT4-induced apoptosis in OSCC.
This study also revealed that invasion of OSCC was inhibited via the downregulation of autophagy. This finding is in accordance with previous studies. For example, inhibition of ROS-dependent NUPR1-mediated autophagy antagonizes repeated cadmium exposure-induced OSCC invasion (Fan et al., 2019). Moreover, PI3K inhibitors targeted disruption of PI3K/Akt/mTOR signaling pathway suppress autophagy and decrease the invasiveness of OSCC (Aggarwal et al., 2019). In addition, silencing phosphofructokinase-platelet (PFKP) prohibits starvation-induced autophagy and the subsequent epithelial–mesenchymal transition (EMT) required to invasion in OSCC (Chen et al., 2018). Interestingly, PI3K/AKT signaling pathway contributes to both apoptosis and invasion, suggesting that PI3K/AKT may be one of the subsequent signaling pathways of KRT4 in OSCC tumor progression.
Further analysis demonstrated that KRT4 inhibited ATG4B-mediated autophagy via disrupting the interaction between ATG4B and LC3. ATG4B induces autophagy through interacting with LC3 in several ways. For example, ATG4B hydrolyses LC3 at the C-terminal region to expose a glycine residue to LC3II required for autophagosome formation (Shu et al., 2010; Li et al., 2011). ATG4B is also required for the deconjugation of membrane-bound MAP1LC3-II from non-autophagosomes or from autophagosomes to support the LC3II supplement (Yu et al., 2012). The interaction between ATG4B and LC3 is critical for facilitating autophagy. A previous study has indicated that phosphorylation of ATG4B at Ser383 and Ser392 decreases the proteolytic activity of ATG4B and leads to delipidating MAP1LC3-II (Yang et al., 2015). Thus, this finding suggested that KRT4 may competitively bind with ATG4B to disturb the interaction of ATG4B and LC3.
To date, therapies against OSCC are limited. Cancer stem cells (CSCs) contribute to the metastasis of OSCC through modulating cytokines and regulating cadherins (Najafi et al., 2019). Thus, CSCs may become a promising therapeutic target for OSCC. Autophagy is involved in modulating stemness, recurrence, and resistance to anticancer therapies (Yun and Lee, 2018). However, the role of KRT4 in CSCs of OSCC remains unclear. According to the findings of this study, we guessed KRT4 may inhibit autophagy in CSCs of OSCC and subsequently suppress the metastasis of OSCC, which may be a potential therapeutic drug target for OSCC through regulating CSCs.
Summarily, this study revealed that KRT4 suppressed OSCC development through inhibiting ATG4B-mediated autophagy, which uncovered the cancer-suppressive role of KRT4 for the first time. Moreover, the finding of the present study may provide a potential therapeutic drug target for OSCC.
Availability of Data and Materials: The data of this study are available from the corresponding author upon reasonable request.
Author Contribution: The authors confirm contribution to the paper as follows: study conception and design: Xiaoxu Li. Author, Bin Cheng. Author; data collection: Xiaoxu Li. Author, Yun Wang. Author, Juan Fang. Author, Zhi Wang. Author; analysis and interpretation of results: Xiaoxu Li. Author, Yun Wang. Author, Juan Fang. Author, Zhi Wang. Author, Xiaoan TAO. Author, Juan XIA. Author; draft manuscript preparation: Xiaoxu LI. Author, Bin Cheng. Author. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: Not Applicable.
Funding Statement: This work was supported by the National Natural Science Foundation of China (Grant No. 81500864), Guangzhou Science and Technology Project (Grant No. 201804010040), and Sun Yat-Sen University Young Teacher Cultivation Project (Grant No. 18ykpy29).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Aggarwal S, John S, Sapra L, Sharma SC, Das SN (2019). Targeted disruption of PI3K/Akt/mTOR signaling pathway, via PI3K inhibitors, promotes growth inhibitory effects in oral cancer cells. Cancer Chemotherapy and Pharmacology 83: 451–461. DOI 10.1007/s00280-018-3746-x. [Google Scholar] [CrossRef]
Akin D, Wang SK, Habibzadegah-Tari P, Law B, Ostrov D, Li M, Yin XM, Kim JS, Horenstein N, Dunn W (2014). A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 10: 2021–2035. DOI 10.4161/auto.32229. [Google Scholar] [CrossRef]
Akito T, Masaaki K, Taichi H, Ayako S, Chieko K, Satoshi W, Yoshinobu E, Okio H, Keiji T, Noboru M (2011). Autophagy-deficient mice develop multiple liver tumors. Genes & Development 25: 795–800. DOI 10.1101/gad.2016211. [Google Scholar] [CrossRef]
Amaravadi R, Kimmelman AC, White E (2016). Recent insights into the function of autophagy in cancer. Genes & Development 30: 1913–1930. DOI 10.1101/gad.287524.116. [Google Scholar] [CrossRef]
Argiris A, Karamouzis MV, Raben D, Ferris RL (2008). Head and neck cancer. Lancet 371: 1695–1709. DOI 10.1016/S0140-6736(08)60728-X. [Google Scholar] [CrossRef]
Behrends C, Sowa ME, Gygi SP, Harper JW (2010). Network organization of the human autophagy system. Nature 466: 68–76. DOI 10.1038/nature09204. [Google Scholar] [CrossRef]
Calabrese F, Giacometti C, Beghe B, Rea F, Loy M, Zuin R, Marulli G, Baraldo S, Saetta M, Valente M (2005). Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respiratory Research 6: 1256. DOI 10.1186/1465-9921-6-14. [Google Scholar] [CrossRef]
Chao SC, Tsai YM, Yang MH, Lee JY (2003). A novel mutation in the keratin 4 gene causing white sponge naevus. British Journal of Dermatology 148: 1125–1128. DOI 10.1046/j.1365-2133.2003.05337.x. [Google Scholar] [CrossRef]
Chen G, Liu H, Zhang Y, Liang J, Zhu Y, Zhang M, Yu D, Wang C, Hou J (2018). Silencing PFKP inhibits starvation-induced autophagy, glycolysis, and epithelial mesenchymal transition in oral squamous cell carcinoma. Experimental Cell Research 370: 46–57. DOI 10.1016/j.yexcr.2018.06.007. [Google Scholar] [CrossRef]
Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, Chou SH, Chien CS, Ku HH, Lo JF (2008). Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clinical Cancer Research 14: 4085–4095. DOI 10.1158/1078-0432.CCR-07-4404. [Google Scholar] [CrossRef]
Choi S, Myers JN (2008). Molecular pathogenesis of oral squamous cell carcinoma: Implications for therapy. Journal of Dental Research 87: 14–32. DOI 10.1177/154405910808700104. [Google Scholar] [CrossRef]
Chon JK, Borkowski A, Partin AW, Isaacs JT, Jacobs SC, Kyprianou N (1999). Alpha 1-adrenoceptor antagonists terazosin and doxazosin induce prostate apoptosis without affecting cell proliferation in patients with benign prostatic hyperplasia. Journal of Urology 161: 2002–2008. DOI 10.1016/S0022-5347(05)68873-8. [Google Scholar] [CrossRef]
Ethiraj P, Sambandam Y, Hathaway-Schrader JD, Haque A, Novince CM, Reddy SV (2019). RANKL triggers resistance to TRAIL-induced cell death in oral squamous cell carcinoma. Journal of Cellular Physiology 235: 1663–1673. DOI 10.1002/jcp.29086. [Google Scholar] [CrossRef]
Fan T, Chen Y, He Z, Wang Q, Yang X, Ren Z, Zhang S (2019). Inhibition of ROS/NUPR1-dependent autophagy antagonises repeated cadmium exposure-induced oral squamous cell carcinoma cell migration and invasion. Toxicology Letters 314: 142–152. DOI 10.1016/j.toxlet.2019.07.017. [Google Scholar] [CrossRef]
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (2010). Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International Journal of Cancer 127: 2893–2917. DOI 10.1002/ijc.25516. [Google Scholar] [CrossRef]
Guo, J. Y., Karsli-Uzunbas, G., Mathew, R., Aisner, S. C., Kamphorst, J. J. et al. (2013). Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes & Development 27: 1447–1461. DOI 10.1101/gad.219642.113. [Google Scholar] [CrossRef]
Haddad RI, Shin DM (2008). Recent advances in head and neck cancer. New England Journal of Medicine 359: 1143–1154. DOI 10.1056/NEJMra0707975. [Google Scholar] [CrossRef]
Hashibe M, Brennan P, Chuang SC, Boccia S, Castellsague X, Chen C, Curado MP, Dal Maso L, Daudt AW, Fabianova E, Fernandez L, Wünsch-Filho V, Franceschi S, Hayes RB, Herrero R, Kelsey K, Koifman S, La Vecchia C, Lazarus P, Levi F, Lence JJ, Mates D, Matos E, Menezes A, McClean MD, Muscat J, Eluf-Neto J, Olshan AF, Purdue M, Rudnai P, Schwartz SM, Smith E, Sturgis EM, Szeszenia-Dabrowska N, Talamini R, Wei Q, Winn DM, Shangina O, Pilarska A, Zhang ZF, Ferro G, Berthiller J, Boffetta P (2009). Interaction between tobacco and alcohol use and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiology and Prevention Biomarkers 18: 541–550. DOI 10.1158/1055-9965.EPI-08-0347. [Google Scholar] [CrossRef]
Huo, Y., Cai, H., Teplova, I., Bowman-Colin, C., Chen, G. et al. (2013). Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discovery 3: 894–907. DOI 10.1158/2159-8290.CD-13-0011. [Google Scholar] [CrossRef]
Kademani D (2007). Oral cancer. Mayo Clinic Proceedings 82: 878–887. DOI 10.4065/82.7.878. [Google Scholar] [CrossRef]
Kimura M, Nagao T, Machida J, Warnakulasuriya S (2013). Mutation of keratin 4 gene causing white sponge nevus in a Japanese family. International Journal of Oral and Maxillofacial Surgery 42: 615–618. DOI 10.1016/j.ijom.2012.10.030. [Google Scholar] [CrossRef]
Lallemant B, Evrard A, Combescure C, Chapuis H, Chambon G, Raynal C, Reynaud C, Sabra O, Joubert D, Hollande F, Lallemant J, Lumbroso S, Brouillet J (2009). Clinical relevance of nine transcriptional molecular markers for the diagnosis of head and neck squamous cell carcinoma in tissue and saliva rinse. BMC Cancer 9: 581. DOI 10.1186/1471-2407-9-370. [Google Scholar] [CrossRef]
Levine B (2007). Autophagy and cancer. Nature 446: 745–747. DOI 10.1038/446745a. [Google Scholar] [CrossRef]
Levine B, Kroemer G (2019). Biological functions of autophagy genes: a disease perspective. Cell 176: 11–42. DOI 10.1016/j.cell.2018.09.048. [Google Scholar] [CrossRef]
Li B, Lu M, Jiang XX, Pan MX, Mao JW, Chen M (2017). Inhibiting reactive oxygen species-dependent autophagy enhanced baicalein-induced apoptosis in oral squamous cell carcinoma. Journal of Natural Medicines 71: 433–441. DOI 10.1007/s11418-017-1076-7. [Google Scholar] [CrossRef]
Lo WL, Kao SY, Chi LY, Wong YK, Chang RC (2003). Outcomes of oral squamous cell carcinoma in Taiwan after surgical therapy: Factors affecting survival. Journal of Oral and Maxillofacial Surgery 61: 751–758. DOI 10.1016/S0278-2391(03)00149-6. [Google Scholar] [CrossRef]
Li, M., Hou, Y. F., Wang, J. S., Chen, X. Y., Shao, Z. M., Yin, X. M (2011). Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. Journal of Biological Chemistry 286: 7327–7338. DOI 10.1074/jbc.M110.199059. [Google Scholar] [CrossRef]
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008). Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075. DOI 10.1038/nature06639. [Google Scholar] [CrossRef]
Najafi M, Mortezaee K, Majidpoor J (2019). Cancer stem cell (CSC) resistance drivers. Life Sciences 234: 116781. DOI 10.1016/j.lfs.2019.116781. [Google Scholar] [CrossRef]
Nakatogawa H, Ichimura Y, Ohsumi Y (2007). Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130: 165–178. DOI 10.1016/j.cell.2007.05.021. [Google Scholar] [CrossRef]
Pengo N, Agrotis A, Prak K, Jones J, Ketteler R (2017). A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nature Communications 8: 9. DOI 10.1038/s41467-017-00303-2. [Google Scholar] [CrossRef]
Rosenfeld MR, Xiaobu Y, Supko JG, Serena D, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, McAfee Q, Fisher J, Troxel AB, Piao SF, Heitjan DF, Tan KS, Pontiggia L, O’Dwyer PJ, Davis LE, Amaravadi RK (2014). A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10: 1359–1368. DOI 10.4161/auto.28984. [Google Scholar] [CrossRef]
Shu, C. W., Drag, M., Bekes, M., Zhai, D., Salvesen, G. S., Reed, J. C (2014). Synthetic substrates for measuring activity of autophagy proteases-autophagins (Atg4). Autophagy 6: 936–947. DOI 10.4161/auto.6.7.13075. [Google Scholar] [CrossRef]
Sophia J, Kowshik J, Dwivedi A, Bhutia SK, Manavathi B, Mishra R, Nagini S (2018). Nimbolide, a neem limonoid inhibits cytoprotective autophagy to activate apoptosis via modulation of the PI3K/Akt/GSK-3beta signalling pathway in oral cancer. Cell Death & Disease 9: E359. DOI 10.1038/s41419-018-1126-4. [Google Scholar] [CrossRef]
Takikita M, Hu N, Shou JZ, Giffen C, Wang QH, Wang C, Hewitt SM, Taylor PR (2011). Fascin and CK4 as biomarkers for esophageal squamous cell carcinoma. Anticancer Research 31: 945–952. [Google Scholar]
Tang Z, Takahashi Y (2018). Analysis of protein-protein interaction by Co-IP in human cells. In: Oñate-Sánchez L, (eds.) Two Hybrids Systems. Methods in Molecular Biology. vol. 1794. pp. 289–296. New York, NY: Humana Press. [Google Scholar]
Toruner GA, Ulger C, Alkan M, Galante AT, Rinaggio J, Wilk R, Tian B, Soteropoulos P, Hameed MR, Schwalb MN, Dermody JJ (2004). Association between gene expression profile and tumor invasion in oral squamous cell carcinoma. Cancer Genetics and Cytogenetics 154: 27–35. DOI 10.1016/j.cancergencyto.2004.01.026. [Google Scholar] [CrossRef]
Tran E, Chow A, Goda T, Wong A, Blakely K, Rocha M, Taeb S, Hoang VC, Liu SK, Emmenegger U (2013). Context-dependent role of ATG4B as target for autophagy inhibition in prostate cancer therapy. Biochemical and Biophysical Research Communications 441: 726–731. DOI 10.1016/j.bbrc.2013.10.117. [Google Scholar] [CrossRef]
Weidberg H, Shvets E, Elazar Z (2011). Biogenesis and cargo selectivity of autophagosomes. Annual Review of Biochemistry 80: 125–156. DOI 10.1146/annurev-biochem-052709-094552. [Google Scholar] [CrossRef]
Yang, Z. F., Wilkie-Grantham, R. P., Yanagi, T., Shu, C. W., Matsuzawa, S. I., Reed, J. C. (2015). ATG4B (autophagin-1) phosphorylation modulates autophagy. Journal of Biological Chemistry 290: 26549–26561. DOI 10.1074/jbc.M115.658088. [Google Scholar] [CrossRef]
Ye H, Yu T, Temam S, Ziober BL, Wang J, Schmartz JL, Mao L, Wong DT, Zhou XF (2008). Transcriptomic dissection of tongue squamous cell carcinoma. BMC Genomics 9: 69. DOI 10.1186/1471-2164-9-69. [Google Scholar] [CrossRef]
Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou SS, Chen Y, Zheng XL, Klionsky DJ, Liang YH, Xie ZP (2014). Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy 8: 883–892. DOI 10.4161/auto.19652. [Google Scholar] [CrossRef]
Yun CW, Lee SH (2018). The roles of autophagy in cancer. International Journal of Molecular Sciences 19: 3466. DOI 10.3390/ijms19113466. [Google Scholar] [CrossRef]
Zhang J, Quan J, Ren Y, Chen M, Yang J, Zhang X (2018a). Keratin 4 regulates the development of human white sponge nevus. Journal of Oral Pathology & Medicine 47: 598–605. DOI 10.1111/jop.12728. [Google Scholar] [CrossRef]
Zhang JM, Yang ZW, Chen RY, Gao P, Zhang YR, Zhang LF (2009). Two new mutations in the keratin 4 gene causing oral white sponge nevus in Chinese family. Oral Diseases 15: 100–105. DOI 10.1111/j.1601-0825.2008.01498.x. [Google Scholar] [CrossRef]
Zhang M, Han N, Jiang Y, Wang J, Li G, Lv X, Li G, Qiao Q (2018b). EGFR confers radioresistance in human oropharyngeal carcinoma by activating endoplasmic reticulum stress signaling PERK-eIF2α-GRP94 and IRE1α-XBP1-GRP78. Cancer Medicine 7: 6234–6246. DOI 10.1002/cam4.1862. [Google Scholar] [CrossRef]
Zheng CM, Ge MH, Zhang SS, Tan Z, Wang P, Zheng RS, Chen WQ, Xia QM (2015). Oral cavity cancer incidence and mortality in China, 2010. Journal of Cancer Research and Therapeutics 11: C149–154. [Google Scholar]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |