DOI:10.32604/biocell.2021.013027
| BIOCELL DOI:10.32604/biocell.2021.013027 | |
| Article |
HSA-MIR-203/MyD88 axis mediates the protective effect of hispidulin on LPS-induced apoptosis in a human renal tubular epithelial line, HK-2
1Department of Critical Care Medicine, The Second Affiliated Hospital of Harbin Medical University, Harbin, 150001, China
2Department of Critical Care Medicine, The Cancer Hospital of Harbin Medical University, Harbin, 150081, China
3Department of Critical Care Medicine, The First Affiliated Hospital of Harbin Medical University, Harbin, 150001, China
*Address correspondence to: Kaijiang Yu, kaijiangyu12@163.com
Received: 22 July 2020; Accepted: 21 December 2020
Abstract: Acute kidney injury (AKI), commonly occurring as complications of sepsis, cardiac surgery, and liver or kidney transplantation, is a critical care syndrome. It is well known that lipopolysaccharide (LPS) shock is a common triggering factor for AKI. This study is aimed to examine the effect of flavonoid compound hispidulin on LPS-induced AKI. For this, renal tubular epithelial cell HK-2 was treated with LPS to establish an in vitro model of AKI. The effect of hispidulin on HK-2 cell viability was examined using CCK-8 assay. Cell apoptosis was determined by TUNEL and flow cytometry. Apoptosis marker proteins were determined by using western blot. The levels of pro-inflammatory cytokines were determined by ELISA assay and qRT-PCR. The translocation of NF-κB was determined by western blot. The effect of MyD88 on the cytoprotective activities of hispidulin was examined by overexpressing MyD88 in HK-2 cells. Our results showed that hispidulin was not able to produce a cytotoxic effect on HK-2 cells at tested concentrations. However, hispidulin could protect HK-2 cells from LPS-induced cell injury. Our results also showed that hispidulin was able to attenuate LPS-induced HK-2 cell apoptosis. In addition, LPS led to an inflammatory response in HK-2 cells, evidenced by NF-κB p65 activation as well as increased expression and release of inflammatory cytokine IL-6 and TNF-α, which could be reversed by pretreatment with hispidulin. Overexpression MyD88 was found to significantly dampen the cytoprotective activities of hispidulin against LPS insult. More importantly, MyD88 was identified as a direct target of hsa-miR-203, and hispidulin was found to regulate the expression of MyD88 via upregulating hsa-miR-203. Our results showed that hispidulin attenuates LPS-induced HK-2 damage via regulating hsa-miR-203/MyD88 axis.
Keywords: Hispidulin; LPS; HK-2; hsa-miR-203; MyD88
Acute kidney injury (AKI), featured by a rapid decrease in renal function, has been considered as a critical care syndrome (Kölling et al., 2018). AKI commonly occurs as complications of sepsis, cardiac surgery, and liver or kidney transplantation (Li et al., 2013). According to the statistics, AKI accounts for almost half of death mortality in intensive care unit (ICU) patients (Mccoy et al., 2019). It has been established that the secondary outcome of systemic infections, endotoxic (lipopolysaccharide, LPS) shock, remains the most frequent trigger for AKI. Therefore, the discovery of agents that can alleviate LPS-induced renal injury is necessary (Mccoy et al., 2019).
As a class of non-coding RNA with a length of 21-25 nucleotides, microRNAs (miRNAs) have been found to universally express in human tissues (Li et al., 2020). By post-transcriptionally regulate gene expression by binding to 3’-untranslated regions (3’-UTR) of target mRNAs, miRNAs play a key role in the pathogenesis of a variety of human conditions (Zhang et al., 2019). Interestingly, a number of miRNAs have been found to correlate with AKI. For instance, miRNA-20a has been recently found to suppress LPS-induced HK-2 cell injury (Zhang et al., 2020). MiR-188 has also been reported to augment contrast-induced acute kidney injury in vivo and in vitro (Liu, 2019).
Since the past two decades, the nephroprotective effects of flavonoid compounds have been noted. Ren et al. (2020) have reported that flavonoid fisetin alleviates inflammatory response in a murine model of septic AKI. Luteolin has also been found to exhibit protective activities against LPS-induced AKI in mice model (Xin et al., 2016). As a natural flavonoid, hispidulin has been identified as the active ingredient in a few traditional Chinese medicines such as Artemisia vestita and Arnica montana L. Hispidulin has displayed a variety of pharmacological properties, including anticancer, anti-fungal, anti-diabetic, immunosuppressive, and neuroprotective activities (Thitilertdecha et al., 2019). Moreover, a recent report has documented that hispidulin suppressed mast cell-mediated allergic inflammation in vitro and in vivo (Kim et al., 2019). However, the role of hispidulin in LPS-induced cell injury has never been explored. In this study, renal tubular epithelial cell HK-2 is treated with LPS to establish an in vitro model of AKI. The protective effect of hispidulin against LPS-induced cell injury in HK-2 cells is explored, and the underlying mechanisms are examined.
HK-2 cells were purchased from the American Type Culture Collection (ATCC). Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, Carlsbad, CA) was used as a culture medium. The DMEM medium was supplemented with penicillin and streptomycin (Invitrogen) and 10% fetal bovine serum (Hyclone, Logan, UT). Hispidulin was purchased from Herbpurify Co. Ltd. (Chengdu, China).
Hsa-miR-203 mimic, inhibitor, and the negative control (NC) were synthesized by GenePharma Co. (Shanghai, China). HK-2 cells were transfected with hsa-miR-203 mimic or inhibitor using the Lipofectamine 3000 (Invitrogen, Carlsbad, CA). The sequence of Myeloid differentiation factor88 (MyD88) full-length cDNA was ligated into the pEX-2 plasmid (GenePharma, Shanghai, China), referring to MyD88 DNA. Cells were transfected with pEX-MyD88 through the Lipofectamine 3000 (Invitrogen, Carlsbad, CA).
Cell counting kit-8 (CCK-8) assay
Cell viability was assessed using a CCK-8 kit (Beyotime, Shanghai, China) in accordance with the manufacturer’s instructions. After treatment, absorbance was detected at 450 nm using a spectrophotometer (Tecan Group Ltd., Männedorf, Switzerland) to assess proliferation.
qRT-PCR (quantitative real-time polymerase chain reaction) analysis
Total RNA was extracted from cells using Trizol reagent (Life Technologies, USA). Thereafter, TaqMan Reverse Transcription Reagents (Applied Biosystems, USA) were employed to reverse transcription of total RNA into complementary DNA (cDNA). The SYBR Green PCR Master Mix (Takara, Japan) was used to quantify the expression levels of targeted genes at the transcriptional level. The sequences of the primers for hsa-miR-203 detection were designed and synthesized as follows: forward primer 5’-AGTGGTTCTTGACAGTTCAACA-3’; reverse primer 5’-GCTGTCAACGATACGCTACGTAACG-3’. The sequences of the primers for MyD88 detection were designed and synthesized as follows: forward primer 5’-CATACCCTTGGTCGCGCTTA-3’; reverse primer 5’-TCCGAGGGTTCAAGAACAGC-3’. The sequences of the primers for IL-6 detection were designed and synthesized as follows: forward primer 5’-TCCAAGACCAGGGTGGCTGGGAAG-3’; reverse primer 5’-TGCCATGTGGGGGTCCCGAA-3’. The sequences of the primers for IL-6 detection were designed and synthesized as follows: forward primer 5’-TTTGCTGGGGCTCATTCACT-3’; reverse primer 5’-GACTCGGCACTTAGCACTGT-3’. The 2-ΔΔCt method was used to calculate fold changes between the experiment group and the control group.
The total protein of the cell lines was extracted using Radio-Immunoprecipitation Assay (RIPA) buffer with added Protease Inhibitor Cocktail (Cwbio, China), incubated on ice for 30 min, followed by centrifugation for 15 min at 14000 × g. The nuclear protein was extracted according to the standard protocols. The proteins were separated SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and then transferred onto Polyvinylidene Fluoride (PVDF) membranes (Millipore, USA). Following blockade with nonfat milk, the PVDF membranes were incubated with specific primary antibodies. The membranes were incubated with Horseradish Peroxidase (HRP)-coupled secondary antibody (Abcam, Cambridge, MA) for 1 h at room temperature before being revealed using enhanced chemiluminescence reagent (Beyotime, Shanghai, China).
The luciferase reporter gene assay
The target gene of hsa-miR-203 was analyzed and predicted in the online website (http://www.microrna.org). The MyD88 dual-luciferase reporter gene vector (pmiRRB-MYD88-3’UTR) was constructed by GenePharma Co. (Shanghai, China). HK-2 cells were co-transduced with hsa-miR-203 mimic, hsa-miR-203 inhibitor, or NC, respectively. 48 h following transfection, the luciferase activity was detected using a dual-luciferase reporter gene detection system (Promega, Madison, WI).
Enzyme-linked immunosorbent assay (ELISA)
Samples from treated and non-treated HK-2 cells were centrifuged, and supernatants were collected for quantification. Tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) levels were quantified by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Tunel (terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end Labeling assay)
Cell apoptosis rates were analyzed by using the TUNEL Cell Apoptosis Assay Kit (Solarbio, Beijing, China) according to the manufacturer’s protocol. Briefly, cells were double-stained with TUNEL staining and 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) for 30 min, then cells were washed with phosphate-buffered saline (PBS) for 3 times and resuspended in 500 μL PBS.
Apoptosis was detected using Annexin V-fluoresceine isothiocyanate (FITC) apoptosis kit (Cell Signaling Technology, Danvers, MA). Briefly, the cells were digested using trypsin before stained in Annexin V-FITC and propidium iodide (PI) for 30 min in the dark. Then, cell apoptosis was detected using a flow cytometer (Thermo Fisher Scientific, Waltham, MA).
Experiments were performed in triplicate and repeated three times. All data are presented as the mean ± standard deviation. The statistical software SPSS 20.0 was used for data analysis, and relevant images were drawn by GraphPad Prism Software (Version X, La Jolla, CA). Differences between two groups were analyzed using an unpaired Students’ t-test. Differences among multiple groups were analyzed using one-way ANOVA. A P-value < 0.05 was considered statistically significant.
Hispidulin rescues HK-2 cells from LPS-induced injury
The chemical formula is presented in Fig. 1A. At first, we examined whether hispidulin treatment affected the viability of HK-2 cells. HK-2 cells were incubated with hispidulin at 2, 5, and 10 μM for 24 h, and the cell viability was assessed via CCK-8 assay. As shown in Fig. 1B, hispidulin at 2, 5, and 10 μM did not markedly change the cell viability of HK-2 cells. Next, we examined whether hispidulin was able to rescue HK-2 cells from LPS-caused cell injury. HK-2 cells were treated with LPS at 2 μg/mL for 12 h, and our results showed that the percentage of viable cells dropped to less than 50% (Fig. 1C). Then HK-2 cells were pretreated with hispidulin at indicated concentrations for 24 h before challenged with LPS at 2 μg/mL for 12 h. Hispidulin at 2 μM failed to attenuate LPS-induced cell injury. In contrast, hispidulin at 5 μM effectively increased the cell viability to more than 75% (Fig. 1C). In the case of 10 μM, hispidulin increased the cell viability to approximately 85% (Fig. 1C). Next, we examined whether hispidulin treatment was able to protect LPS-induce apoptosis in HK-2 cells. Apoptosis was assessed firstly by TUNEL assay. As shown in Fig. 1D, LPS treatment led to a marked increase in the TUNEL-positive population. In contrast, pretreatment with hispidulin at 5 and 10 μM was able to significantly reduce the proportion of TUNEL-positive cells. Cell apoptosis was also detected using flow cytometry, which also showed that LPS increased cell apoptosis to more than 15% (Fig. 1E). Hispidulin at 5 μM effectively reduced the cell apoptosis to approximately 12% (Fig. 1E). In the case of 10 μM, hispidulin further reduced cell apoptosis to less than 10% (Fig. 1E). Activation of caspase-3 is a hallmark of cell apoptosis. In our study, we found that LPS treatment led to a significant increase in the level of cleaved caspase-3 (Fig. 1F). In contrast, hispidulin at both 5 and 10 μM was able to reduce LPS-induced elevation of cleaved caspase-3 level (Fig. 1F). It has been found that LPS was able to regulate the expression of anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax in HK-2 cells (Wang et al., 2018). In agreement with these results, our results showed that LPS downregulated Bcl-2 while upregulated Bax (Fig. 1F). In contrast, our results showed that hispidulin was able to markedly attenuate LPS-triggered caspase-3 cleavage, Bcl-2 downregulation, and Bax upregulation (Fig. 1F).

Figure 1: Hispidulin rescued HK-2 from LPS-induced cell injury.
(A) The chemical structure of hispidulin was presented. (B) HK-2 cells were treated with hispidulin for 24 h at the indicated concentration, and the cell viability was determined using CCK-8 assay. (C–E) HK-2 cells were pretreated with hispidulin at the indicated concentration for 24 h and then stimulated with LPS at 2 μg/mL for 12 h. (C) The cell viability was determined by CCK-8 assay. (D) Cell apoptosis was assessed by TUNEL assay. (E) Cell apoptosis was assessed by flow cytometry. (F) The expression levels of cleaved caspase-3, Bax, and Bcl-2 were assessed by western blot. **P < 0.01.
Hispidulin alleviates HK-2 cells from LPS-induced inflammatory response
Then the effect of hispidulin on LPS-induced response in HK-2 cells was examined. As shown in Fig. 2A, the mRNA level of IL-6 was increased by LPS treatment to more than 200% compared with control. The mRNA of TNF-α was increased by LPS treatment to more than 500% compared with control (Fig. 2A). Then HK-2 cells were pretreated with hispidulin at indicated concentrations for 24 h before challenged with LPS at 2 μg/mL for 12 h. Our results showed that hispidulin at 5 and 10 μM was capable of reducing the mRNA level of IL-6 and TNF-α in HK-2 cells in a dose-dependent manner (Fig. 2A). Correspondingly, our findings revealed that LPS was able to elevate the protein expression of both IL-6 and TNF-α, which could be reversed by pretreatment with hispidulin (Fig. 2B). The levels of released cytokines in the supernatant were also detected using ELISA assay. As shown in Fig. 2C, LPS treatment increased the level of IL-6 in supernatant from less than 30 pg/mL to about 200 pg/mL. In contrast, hispidulin was capable of reducing LPS-induced elevation in IL-6 release in a dose-dependent manner (Fig. 2C). LPS treatment increased the level of TNF-α in the supernatant to more than 50 pg/mL, while hispidulin at 5 and 10 μM markedly reduced the level of TNF-α (Fig. 2C). Moreover, our results showed that LPS promoted the translocation of p65 into the nucleus, which was partially abrogated by pretreatment of hispidulin (Fig. 2D). All these data pointed out that hispidulin alleviates LPS-induced inflammatory response in HK-2 cells.
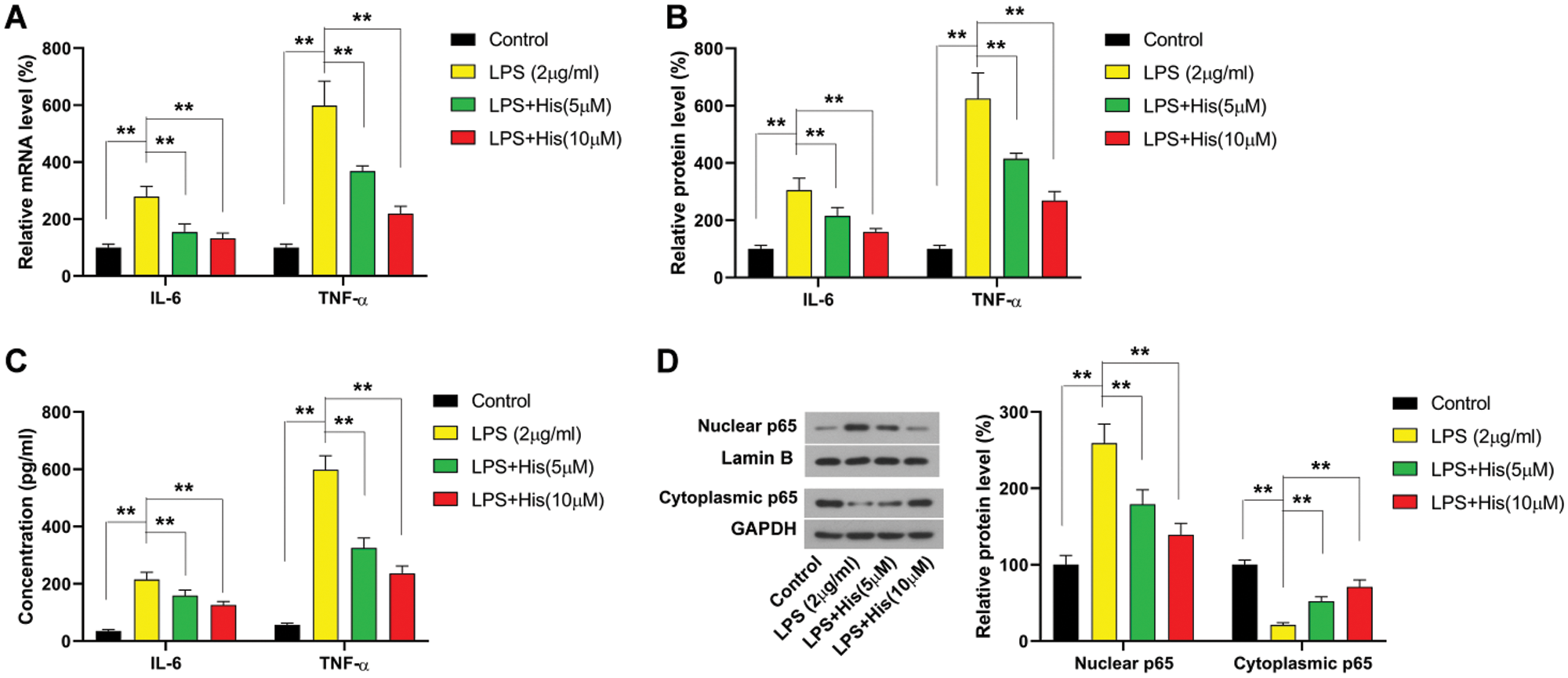
Figure 2: Hispidulin ameliorates LPS-induced inflammatory response in HK-2 cells. HK-2 cells were pretreated with hispidulin at the indicated concentration for 24 h and then stimulated with LPS at 2 μg/mL for 12 h.
(A) The mRNA expression level of TNF-α and IL-6 was assessed by qRT-PCR. (B) The protein expression level of TNF-α and IL-6 was assessed by western blot. (C) The levels of TNF-α and IL-6 in the supernatant were assessed by ELISA assay. (D) The protein expression levels of nuclear p65 and cytoplasmic p65 were assessed by western blot. **P < 0.01.
Hispidulin protects HK-2 cells from LPS-induced cell injury in MyD88-dependent manner
Next, we examine the involvement of MyD88 in the protective effect of hispidulin against LPS-induced cell injury in HK-2 cells. HK-2 cells were pretreated with hispidulin at indicated concentrations for 24 h before challenged with LPS at 2 μg/mL for 12 h. Consistent with previous studies, LPS treatment led to a significant increase in the protein expression of MyD88 (Fig. 3A). In contrast, pretreatment with hispidulin was able to downregulate MyD88 in LPS-treated HK-2 cells (Fig. 3A). Then MyD88 expression was elevated by transfecting HK-2 cells with MyD88 overexpressing vector. 48 h post-transfection, HK-2 cells were pretreated with hispidulin at indicated concentrations for 24 h before challenged with LPS at 2 μg/mL for 12 h. Our results showed that in HK-2 cells with ectopic MyD88 expression, pretreatment with hispidulin failed to rescue LPS-induced cell injury (Fig. 3B). Pretreatment with hispidulin reduced LPS-induced apoptosis to approximately 5%, which was totally abolished by ectopic MyD88 expression (Fig. 3C). MyD88 overexpression also completely reversed the effect of hispidulin on LPS-induced caspase-3 activation, Bcl-2 downregulation, and Bax upregulation (Fig. 3D). In addition, we found that the suppressing effect of hispidulin on LPS-induced NF-κB p65 activation was also markedly dampened by ectopic MyD88 expression (Fig. 3D). Moreover, our MyD88 restored the levels of released IL-6 and TNF-α in the supernatant (Fig. 3F). These data showed that the cytoprotective and anti-inflammatory activities of hispidulin depended on its suppression of MyD88.

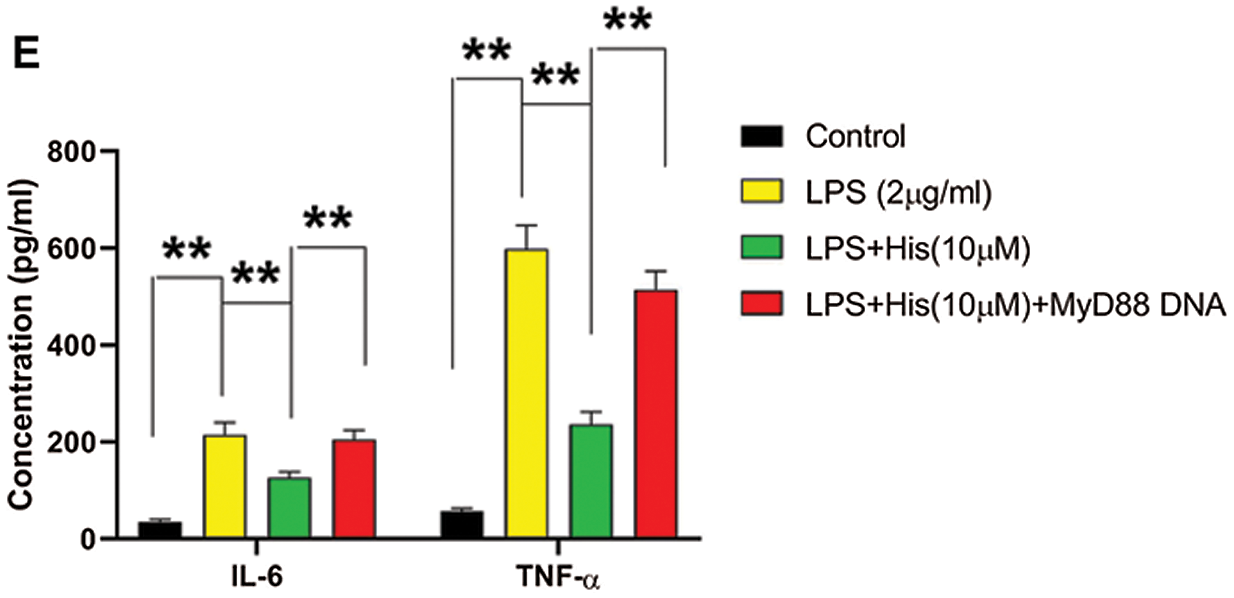
Figure 3: Hispidulin alleviates LPS-induced cell injury in HK-2 cells via downregulating MyD88
(A) HK-2 cells were pretreated with hispidulin at the indicated concentration for 24 h and then stimulated with LPS at 2 μg/mL for 12 h. The protein expression of MyD88 was assessed by western blot. (B–E) HK-2 cells were transfected with MyD88 overexpressing plasmid. 48 h posttransfection, HK-2 cells were pretreated with hispidulin at the indicated concentration for 24 h and then stimulated with LPS at 2 μg/mL for 12 h. B. The cell viability was determined by the CCK-8 assay. (C) Cell apoptosis was assessed by flow cytometry. (D) The protein expression levels of cleaved caspase-3, Bax, Bcl-2, nuclear p65, and cytoplasmic p65 were assessed by western blot. (E) The levels of TNF-α and IL-6 in the supernatant were assessed by ELISA assay. **P < 0.01.
Hispidulin suppresses MyD88 via upregulating hsa-miR-203
The binding sites on the MyD88 sequence with hsa-miR-203 have been predicted by online software, and the mutant sequence has been designed as displayed in Fig. 4A. Then whether MyD88 was a direct target of hsa-miR-203 was validated by luciferase activity assay. As shown in Fig. 4B, hsa-miR-203 mimic significantly decreased the luciferase activities of MyD88 wildtype, while did not markedly affect the luciferase activities of MyD88 with mutant binding sites, which supported that hsa-miR-203 modulate the expression MyD88 by directly binding to its 3’UTR. Then HK-2 cells were transfected with hsa-miR-203 mimic and hsa-miR-203 inhibitor to manipulate the expression of hsa-miR-203. Our results showed that the hsa-miR-203 inhibitor successfully reduced the expression of hsa-miR-203 to less than 40% compared with controls (Fig. 4C). In contrast, hsa-miR-203 mimic effectively elevated the expression of hsa-miR-203 to approximately 2.5-fold compared to that of control (Fig. 4C). Then we examined the effect of hsa-miR-203 inhibitor and hsa-miR-203 mimic on protein expression levels of MyD88. Our results showed that the hsa-miR-203 inhibitor increased the protein level of MyD88 to about 150% of control (Fig. 4D). In contrast, the hsa-miR-203 mimic was able to reduce the protein expression level of MyD88 to less than 50% of control (Fig. 4D). Then the effect of hispidulin on the hsa-miR-203 expression was evaluated. Results from qRT-PCR showed that LPS treatment led to a more than 50% reduction in the level of hsa-miR-203 in HK-2 cells (Fig. 4E). Nevertheless, pretreatment with hispidulin was able to reverse LPS-induced decrease in hsa-miR-203 level (Fig. 4E). Furthermore, our results showed that the hsa-miR-203 inhibitor effectively restored the protein expression level of MyD88 in HK-2 treated with hispidulin and LPS (Fig. 4F). Collectively, this evidence pointed out that hispidulin suppressed MyD88 by upregulating hsa-miR-203.

Figure 4: Hispidulin downregulates MyD88 via increasing the level of hsa-miR-203.
(A) Prediction of MyD88 as a target gene of hsa-miR-203. (B) The relative luciferase activities following transfection were examined using a dual-luciferase reporter gene detection system. (C–D) HK-2 cells were transfected with hsa-miR-203 mimic or hsa-miR-203 inhibitor. (C) 48 h post-transfection, the expression levels of hsa-miR-203 was assessed by qRT-PCR. (D) 48 h post-transfection, the protein expression levels of MyD88 were assessed by western blot. (E) HK-2 cells were pretreated with hispidulin at the indicated concentration for 24 h and then stimulated with LPS at 2 μg/mL for 12 h. The expression levels of hsa-miR-203 were assessed by qRT-PCR. (F) HK-2 cells were transfected with an hsa-miR-203 inhibitor. 48 h post-transfection, HK-2 cells were pretreated with hispidulin at the indicated concentration for 24 h and then stimulated with LPS at 2 μg/mL for 12 h. The protein expression levels were assessed by western blot. **P < 0.01.
Hispidulin exhibits cytoprotective activities via upregulating hsa-miR-203
In the following experiments, we examined the role of hsa-miR-203 in the cytoprotective activities of hispidulin against LPS-induced cell injury. HK-2 cells were transfected with hsa-miR-203 inhibitor and then treated with hispidulin for 24 h before challenge with LPS. Our results showed that the protective effect of hispidulin against LPS-induced cell death was almost completely lost in cells transfected with hsa-miR-203 inhibitor (Fig. 5A). The antagonizing effect of hispidulin against LPS-induced apoptosis was also significantly abolished by the hsa-miR-203 inhibitor (Fig. 5B). As expected, the hsa-miR-203 inhibitor restored the protein levels of cleaved caspase-3 and Bax (Fig. 5C). In contrast, the hsa-miR-203 inhibitor abolished the enhancing effect of hispidulin in the expression of anti-apoptotic protein Bcl-2 (Fig. 5C). Moreover, the hsa-miR-203 inhibitor restored the activity of NF-κB p65 in HK-2 cells treated with both LPS and hispidulin (Fig. 5C). As a consequence of the restoration of NF-κB activities, the suppressing effect of hispidulin on the levels of released cytokines was also abrogated by the hsa-miR-203 inhibitor (Fig. 5D). Taken together, our results showed that hispidulin attenuates LPS-induced HK-2 damage via regulating hsa-miR-203/MyD88 Axis.


Figure 5: Hispidulin alleviates LPS-induced cell injury in HK-2 cells by increasing hsa-miR-203 expression.
HK-2 cells were transfected with an hsa-miR-203 inhibitor. 48 h post-transfection, HK-2 cells were pretreated with hispidulin at the indicated concentration for 24 h and then stimulated with LPS at 2 μg/mL for 12 h. (A) The cell viability was determined by the CCK-8 assay. (B) Cell apoptosis was assessed by flow cytometry. (C) The protein expression levels of cleaved caspase-3, Bax, Bcl-2, nuclear p65, and cytoplasmic p65 were assessed by western blot. (D) The levels of TNF-α and IL-6 in the supernatant were assessed by ELISA assay. **P < 0.01.
AKI, featured by a sudden decline of renal function, increases the chance of development of end-stage renal conditions and also leads to unfavorable clinical outcomes, including death. AKI is associated with a prolonged clinical course due to a number of factors such as toxic or septic insults to the renal parenchyma (Bihorac et al., 2013). The LPS insult, which is a common cause of AKI, could result in renal failure and even multi-organ damage. Therefore, the discovery of agents that can alleviate LPS-induced renal injury is necessary (Jiang et al., 2020). Hispidulin is a naturally occurring flavonoid compound and possesses a variety of pharmacological activities. Recently, it has been reported that hispidulin suppressed mast cell-mediated allergic inflammation in vitro and in vivo (Kim et al., 2019). In the present study, our results showed that hispidulin attenuates LPS-induced HK-2 damage via regulating hsa-miR-203/MyD88 axis.
MyD88 has been identified as a key downstream adaptor for most Toll-like regulators and IL-1 receptors (Zhu et al., 2019). Upon stimulation by LPS, MyD88 is recruited to activate its downstream inflammatory pathways (Zhu et al., 2019). In HK-2 cells, it has been found that LPS-induced MyD88 activation can promote the activation of NF-ĸB and MAPK signaling pathways (Zeng et al., 2020). In fact, it has been found targeting MyD88 provided protection against LPS-induced inflammatory cell injury of HK-2 cells (Guo et al., 2019). In our results, we also evidenced that LPS was able to significantly elevate the expression of MyD88 and activate NF-ĸB signaling, resulting in an inflammatory response in HK-2 cells. Our results also showed that the anti-inflammatory activities of hispidulin were associated with the downregulation of MyD88. To further validate whether MyD88 downregulation mediated the anti-inflammatory reactions, HK-2 cells were transfected with MyD88 overexpressing plasmid. We found that ectopic MyD88 expression restored NF-ĸB activation and the levels of released IL-6 and TNF-α in the supernatant, suggesting that hispidulin protected against LPS-induced inflammatory cell injury by downregulating MyD88.
MiRNAs play key roles in a variety of cellular activities, including inflammatory responses (Li et al., 2020). Moreover, a number of miRNAs have been found to directly target MyD88 upon LPS stimulation, including miR-149, hsa-miR-203, miR-214 (Cui et al., 2019; Zheng et al., 2019). Hsa-miR-203 is reported to be involved in the regulation of inflammatory responses in ischemia (Chen et al., 2017). Hsa-miR-203 was also involved in regulating LPS-induced inflammation in macrophage RAW264.7 cells (Wei et al., 2013). In this study, we focused on the role of hsa-miR-203 in the cytoprotective activities of hispidulin. The data in the present study validated that MyD88 is a direct target gene of hsa-miR-203. Meanwhile, our results showed that hispidulin was able to increase the level of hsa-miR-203 in LPS-treated HK-2 cells. Hsa-miR-203 inhibitor effectively restored the protein expression level of MyD88 in HK-2 treated with hispidulin and LPS. All these pieces of evidence pointed out that hispidulin downregulated MyD88 by increasing the expression of hsa-miR-203. More importantly, the cytoprotective and anti-inflammatory activities of hispidulin against LPS insult almost completely diminished in HK-2 cells when hsa-miR-203 was knockdown. Our results showed that hispidulin attenuates LPS-induced HK-2 damage via regulating hsa-miR-203/MyD88 axis.
This present study showed that hispidulin is able to alleviate LPS-triggered inflammatory cell damage in HK-2 cells, as demonstrated by an increased proportion of viable cells, a decreased proportion of apoptotic cells, and a reduction in production and release of inflammatory cytokines IL-6 and TNF-α. We also explored the relevant molecular mechanisms were also explored, which showed that hispidulin exhibited its cytoprotective activities by targeting the hsa-miR-203/MyD88 axis. These findings highlighted the cytoprotective action of hispidulin in HK-2 cells, which may provide a preclinical basis for further study.
Authors’ Contributions: The authors confirm contribution to the paper as follows: Study conception and design: Sicong Wang, Kaijiang Yu; data collection: Sicong Wang, Ruijin Liu, Qiuyuan Han; analysis and interpretation of results: Sicong Wang, Ruijin Liu; draft manuscript preparation: Sicong Wang, Ruijin Liu, Qiuyuan Han, Kaijiang Yu. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.
Funding Statement: The authors received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Bihorac A, Baslanti TO, Cuenca AG, Hobson CE, Ang D, Efron PA, Maier RV, Moore FA, Moldawer LL (2013). Acute kidney injury is associated with early cytokine changes after trauma. Journal of Trauma and Acute Care Surgery 74: 1005–1013. DOI 10.1097/TA.0b013e31828586ec. [Google Scholar] [CrossRef]
Cui S, Tang J, Wang S, Li L (2019). Kaempferol protects lipopolysaccharide-induced inflammatory injury in human aortic endothelial cells (HAECs) by regulation of miR-203. Biomedicine & Pharmacotherapy 115: 108888. DOI 10.1016/j.biopha.2019.108888. [Google Scholar] [CrossRef]
Chen SJ, Wu P, Sun LJ, Zhou B, Niu W, Liu S, Lin FJ, Jiang GR (2017). miR-204 regulates epithelial-mesenchymal transition by targeting SP1 in the tubular epithelial cells after acute kidney injury induced by ischemia-reperfusion. Oncology Reports 37: 1148–1158. DOI 10.3892/or.2016.5294. [Google Scholar] [CrossRef]
Guo S, Sun J, Zhuang Y (2019). Quercetin alleviates lipopolysaccharide-induced inflammatory responses by up-regulation miR-124 in human renal tubular epithelial cell line HK-2. BioFactors 46: 402–410. DOI 10.1002/biof.1596. [Google Scholar] [CrossRef]
Jiang G, Xin R, Yuan W, Zhang L, Meng X, Sun W, Han H, Hou Y, Wang L, Du P (2020). Ligustrazine ameliorates acute kidney injury through downregulation of NOD2-mediated inflammation. International Journal of Molecular Medicine 45: 731–742. DOI 10.3892/ijmm.2020.4464. [Google Scholar] [CrossRef]
Kim DE, Min KJ, Kim MJ, Kim SH, Kwon TK (2019). Hispidulin inhibits mast cell-mediated allergic inflammation through down-regulation of histamine release and inflammatory cytokines. Molecules 24: 2131. DOI 10.3390/molecules24112131. [Google Scholar] [CrossRef]
Kölling M, Seeger H, Haddad G, Kistler A, Nowak A, Faulhaber-Walter R, Kielstein J, Haller H, Fliser D, Mueller T, Wüthrich RP, Lorenzen JM (2018). The circular RNA ciRs-126 predicts survival in critically ill patients with acute kidney injury. Kidney International Reports 3: 1144–1152. DOI 10.1016/j.ekir.2018.05.012. [Google Scholar] [CrossRef]
Li D, Li G, Chen Y, Li Y, Zhang J, Gao D, Sun L, Liu B (2020). Astragaloside IV protects ATDC5 cells from lipopolysaccharide-caused damage through regulating miR-203/MyD88. Pharmaceutical Biology 58: 89–97. DOI 10.1080/13880209.2019.1705355. [Google Scholar] [CrossRef]
Li PK, Burdmann EA, Mehta RL (2013). World Kidney Day 2013: Acute kidney injury-global health alert. American Journal of Kidney Diseases 61: 359–363. DOI 10.1053/j.ajkd.2013.01.002. [Google Scholar] [CrossRef]
Liu T (2019). Regulation of inflammasome by autophagy, autophagy regulation of innate immunity. In: Cui J, (eds.) Advances in Experimental Medicine and Biology. vol. 1209, pp. 109–123. Singapore: Springer. [Google Scholar]
Mccoy IE, Montez-Rath ME, Chertow GM, Chang TI (2019). Central venous pressure and the risk of diuretic-associated acute kidney injury in patients after cardiac surgery. American Heart Journal 221: 67–73. DOI 10.1016/j.ahj.2019.12.013. [Google Scholar] [CrossRef]
Ren Q, Guo F, Tao S, Huang R, Ma L, Fu P (2020). Flavonoid fisetin alleviates kidney inflammation and apoptosis via inhibiting Src-mediated NF-κB p65 and MAPK signaling pathways in septic AKI mice. Biomedicine & Pharmacotherapy 122: 109772. DOI 10.1016/j.biopha.2019.109772. [Google Scholar] [CrossRef]
Thitilertdecha P, Pluangnooch P, Timalsena S, Soontrapa K (2019). Immunosuppressive effect of hispidulin in allergic contact dermatitis. BMC Complementary and Alternative Medicine 19: 268. DOI 10.1186/s12906-019-2689-z. [Google Scholar] [CrossRef]
Wang X, Ma T, Wan X, Meng Y, Zhao Z, Bian J, Bao R, Deng X, Yang T (2018). IGFBP7 regulates sepsis-induced acute kidney injury through ERK1/2 signaling. Journal of Cell Biochemistry 120: 7602–7611. DOI 10.1002/jcb.28035. [Google Scholar] [CrossRef]
Wei J, Huang X, Zhang Z, Jia W, Zhao Z, Zhang Y, Liu X, Xu G (2013). MyD88 as a target of microRNA-203 in regulation of lipopolysaccharide or Bacille Calmette-Guerin induced inflammatory response of macrophage RAW264.7 cells. Molecular Immunology 55: 303–309. DOI 10.1016/j.molimm.2013.03.004. [Google Scholar] [CrossRef]
Xin SB, Yan H, Ma J, Sun Q, Shen L (2016). Protective effects of luteolin on lipopolysaccharide-induced acute renal injury in mice. Medical Science Monitor 22: 5173–5180. DOI 10.12659/MSM.898177. [Google Scholar] [CrossRef]
Zeng M, Qi M, Wang Y, Xu R, Wu Y, Li M, Zheng X, Feng W (2020). 5-O-methyldihydroquercetin and cilicicone B isolated from Spina Gleditsiae ameliorate lipopolysaccharide-induced acute kidney injury in mice by inhibiting inflammation and oxidative stress via the TLR4/MyD88/TRIF/NLRP3 signaling pathway. International Immunopharmacology 80: 106194. DOI 10.1016/j.intimp.2020.106194. [Google Scholar] [CrossRef]
Zhang B, Shetti D, Fan C, Wei K (2019). miR-29b-3p promotes progression of MDA-MB-231 triple-negative breast cancer cells through downregulating TRAF3. Biological Research 52: 38. DOI 10.1186/s40659-019-0245-4. [Google Scholar] [CrossRef]
Zhang L, He S, Wang Y, Zhu X, Shao W, Xu Q, Cui Z (2020). miRNA-20a suppressed lipopolysaccharide-induced HK-2 cells injury via NFκB and ERK1/2 signaling by targeting CXCL12. Molecular Immunology 118: 117–123. DOI 10.1016/j.molimm.2019.12.009. [Google Scholar] [CrossRef]
Zheng D, Zang Y, Xu H, Wang Y, Cao X, Wang T, Pan M, Shi J, Li X (2019). MicroRNA-214 promotes the calcification of human aortic valve interstitial cells through the acceleration of inflammatory reactions with activated MyD88/NF-kappaB signaling. Clinical Research in Cardiology 108: 691–702. DOI 10.1007/s00392-018-1398-9. [Google Scholar] [CrossRef]
Zhu G, Cheng Z, Lin C, Hoffman RM, Huang Y, Singh SR, Zheng W, Yang S, Ye J (2019). MyD88 regulates LPS-induced NF-κB/MAPK cytokines and promotes inflammation and malignancy in colorectal cancer cells. Cancer Genomics Proteomics 16: 409–419. DOI 10.21873/cgp.20145. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |