DOI:10.32604/biocell.2021.015464
| BIOCELL DOI:10.32604/biocell.2021.015464 | |
| Article |
Exendin-4 inhibits the survival and invasiveness of two colorectal cancer cell lines via suppressing GS3Kβ/β-catenin/NF-κB axis through activating SIRT1
1Biology Department, College of Science, King Khalid University, Abha, 61413, Saudi Arabia
2Zoology Department, College of Science, Damanhour University, Damanhour, 22511, Egypt
3Pathology Department, College of Medicine, Taif University, Taif, 11099, Saudi Arabia
4Biology Department, College of Science for Girls, King Khalid University, Abha, 61413, Saudi Arabia
5Biology Department, College of Science, Princess Nourah Bint Abdulrahman University, Riyadh, 84428, Saudi Arabia
6Biology Department, Faculty of Science and Arts, Dahran Aljnoub, King Khalid University, Abha, 61413, Saudi Arabia
7Research Center for Advanced Materials Science, King Khalid University, Abha, 61413, Saudi Arabia
8Agriculture Research Centre, Soil, Water and Environment Research Institute, Giza, 12619, Egypt
9Mansoura Research Centre for Cord Stem Cell (MARC-CSC), Stem Cells Bank, Children’s Hospital, Mansoura University, Mansoura, 35516, Egypt
*Address correspondence to: Attalla F. El-Kott, elkottaf@kku.edu.sa
Received: 20 December 2020; Accepted: 16 March 2021
Abstract: This study examined if the anti-tumorigenesis effect of Exendin-4 in HT29 and HCT116 colorectal cancer (CRC) involves modulation of SIRT1 and Akt/GSR3K/β-catenin/NF-κB axis. HT29 and HCT116 cells were treated either with increasing levels of Exendin-4 (0.0-200 µM) or with Exendin-4 (at its IC50) in the presence or absence of EX-527 (10 µM/a selective SIRT1 inhibitor) or Exendin-4 (9-39) amide (E (9-39) A) (1 µM/an Exendin-4 antagonist). In a dose-dependent manner, Exendin-4 inhibited cell survival, but enhanced levels of lactate dehydrogenase (LDH) and single-stranded DNA (ssDNA) in both HT29 and HCT116. In both cell lines and at it has an IC50 (45 µM for HT29 and 35 µM for HCT1165), Exendin-4 also significantly reduced cell survival, migration, and invasion of both cell types, with no effect on the expression GLP-1 receptors (GLPRs) nor of the activity of Akt. At these doses, Exendin-4 also increased the expression of SIRT1 but reduced the acetylation of NF-κB and the expression of Bax and cleaved caspase-3 and in both cell lines. Concomitantly, protein levels of p-GS3Kβ (Ser9), total and acetylated β-catenin, and Anix2 were significantly decreased, but levels of p-GS3Kβ (Ser9) and p-β-catenin (Ser33/37/Thr41) were significantly increased in both HT29 and HCT116-exendin-4 treated cells. All the effects exerted by Exendin-4 were completely prevented by Ex527 or E (9-39) A. In conclusion, Exendin-4 suppresses the tumorigenesis of HT29 and HCT116 CRC cell activation of GS3Kβ-induced inhibition of β-catenin and NF-κβ in a SIRT1-dependent mechanism.
Keywords: Colorectal cancer; Exendin-4; SIRT1; GS3Kβ; β-catenin; NF-κB
Colorectal cancer (CRC) is one of the most common solid tumors among male and female patients, which develops due to environmental, nutritional, and genetic factors (Thanikachalam and Khan, 2019). Despite the large improvement in the screening tools and therapeutic options, the mortality rates among the affected patients remained significantly high and are expected to increase to 60% by 2030, with a projection that 90% of those patients will be among the young whose age is between 24–34 years (Siegel et al., 2019).
The highly conserved canonical Wingless/β-catenin (Wnt/β-catenin) signaling pathway is a leading pathway that mainly involved in embryonic development and adult homeostasis, including wound healing, maintenance and renewal of stem cell, and cell proliferation, survival, and motility (El-Sahli et al., 2019; Logan and Nusse, 2004). Under normal conditions, the cytoplasmic levels of β-catenin, a transcription co-activator, remain very low due to their rapid ubiquitination and proteasome degradation as they are always phosphorylated by the β-catenin destruction complex (Krishnamurthy and Kurzrock, 2018). This complex is composed of Serval proteins, including oxen, casein kinase I (CKI), adenoma polyposis coli (APC), and glycogen-synthase kinase-3β (GS3Kβ), where the activation of the GS3Kβ is indispensable for β-catenin-induced degradation (Metcalfe and Bienz, 2011). However, the unphosphorylated β-catenin will escape this degradation and accumulates in the cytoplasm and nucleus to trigger the transcriptional program (Metcalfe and Bienz, 2011). Within this view, the binding of the Wnt protein to its specific transmembrane receptor (Fzd and LRP) triggers the recruitment of Dishevelled proteins, which leads to several events which end up with phosphorylation (inhibition) and/or internalization of GS3Kβ, thus blocking the GS3Kβ-induced β-catenin degradation (Metcalfe and Bienz, 2011; Rajkumar, 2016).
Currently, accumulating evidence is showing that sustained activation of Wnt/β-catenin pathway underlies multiple hematological and solid tumors, including chronic myelogenous leukemia (CML), CRC, and breast, ovaries, prostate, stomach, liver, lung, and thyroid cancers (Beurel and Jope, 2006; El-Sahli et al., 2019; Polakis, 2012; Taciak et al., 2018). Accordingly, the aberrant activation of β-catenin triggers the transcription of numerous genes involved tumor proliferation (e.g., Cyc D1, and Myc), differentiation (e.g., SOX9), stemness (e.g., Nanog and OCT4), and migration (CXCR4, CCL18, ICAM-1, and VCAM-1) (El-Sahli et al., 2019; Polakis, 2012; Rajkumar, 2016). Besides, β-catenin stimulates tumor growth and development by stimulating cancer metabolism, including glycolysis, glutaminolysis, and lipogenesis (El-Sahli et al., 2019). Furthermore, β-catenin can stimulate the tumor inflammatory microenvironment, which plays a significant role in the tumor cells, proliferation, invasiveness, and resistance to drugs by increasing the transcription of TNFSF9 and the nuclear factor-κB (NF-κB) (Hanahan and Weinberg, 2011; Polakis, 2012; Rajkumar, 2016).
However, the process of β-catenin activation seems to be very complicated and involved not Wnt1-dependent mediators. Although it is a major mechanism to induce cancer, inflammation, the components of the NF-κB signaling pathways, including IKKα, RelA (p65), p50, and other inflammatory cytokines, such as IL-6, can upregulate β-catenin transcriptional activity (Albanese et al., 2003; Carayol and Wang, 2006; Cho et al., 2008; Li et al., 2014; Rajkumar, 2016; Schwitalla et al., 2013; Yang et al., 2012). Besides, β-catenin activity is negatively regulated by deacetylation by the silent information regulator 1 (SIRT1) (Cho et al., 2012; Lin and Fang, 2013; Salaroli et al., 2008). However, the contribution of these pathways in β-catenin activation in the different tumor types is still not completely understood.
On the other hand, accumulating line of evidence is currently showing that genetic deletion and silencing or pharmacological inhibition of β-catenin is considered a highly valuable clinical therapeutic option to prevent and slow the progression of CRC (Cong et al., 2003; Green et al., 2001; Mologni et al., 2010; Morin et al., 1996; Polakis, 2012; Tetsu and Mccormick, 1999; van de Wetering et al., 2002; Verma et al., 2003). The glucagon-like peptide-1 analog, Exendin-4, has received much attention during the last years as a therapeutic and anti-tumor drug due to its potent antioxidant and anti-inflammatory potentials, in vivo and in vitro (Defronzo et al., 2005; Lonborg et al., 2014; Yamamoto et al., 2013). Indeed, Exendin-4 anti-tumor effects were demonstrated against breast, prostate, colon, and ovarian cancers through suppressing inflammation and cell survival by modulating several cellular pathways, including NF-κB, PI3K/Akt, AMPK, and MAPKs (Iwaya et al., 2017; Nomiyama et al., 2014; Tsutsumi et al., 2015; Tzotzas et al., 2017). Of note, Exendin-4 also induced cell apoptosis and decreased colony formation CT26 colon cancer cells by increasing intracellular levels of cAMP and inhibiting GS3Kβ and ERK1/2 signaling (Koehler et al., 2011).
Given the complexity of β-catenin activation and its role in CRC, in this study, we hypothesized that Exendin-4 may induce cell death and inhibit cell proliferation and invasiveness of two colorectal cell lines by attenuating the aberrant β-catenin activation through affecting the expression/activities of Akt, GS3Kβ, NF-κB, and SIRT1.
The Human HT29 & HCT116 CRC cell lines were supplied from the American Type Culture (USA) and always cultured in a humidified atmosphere (5% CO2/37°C) in DMEM medium (ThermoFisher Scientific) supplemented with 10% heat-inactivated FBS, 1% penicillin, and 0.1% streptomycin (ThermoFisher Scientific). In all the parts of this study, the cells were always used when they reveal a confluent density of <85%. Accordingly, both cell lines 5 × 104 cells (150 µL cell suspension in the media) were seeded in 96-well tissue culture plates and incubated under standard culture conditions (5% CO2/37°C) for 24 h. each part of any experiment performed in this study was conducted in three experiments each done in triplicate.
Cells were treated with increasing levels of Exendin-4 (10, 25, 50, 100, and 200 µM) (Cat. No. E7144, Sigma Aldrich, MA, USA) for 24 h at 37°C and 5% CO2. Control cells were treated only with 0.05% DMSO (dimethyl sulfoxide) (Cat. No. 276855, Sigma Aldrich, MA, USA). Cell survival, medium levels of lactate dehydrogenase (LDH), and single-stranded DNA (ssDNA) were measured as described below. Based on these data, the inhibitory concentration (IC50) of Extendin-4 in both cell lines was calculated from the survival data and then used for the rest of the experiments. In these experiments, the cells were seeded in the same manner but were incubated with the IC50 of Exendin-4 (45 µM for HT29 and 35 µM for HCT1165). In some cases, cells of both cell lines were also pre-incubated for 1 h with 10 µM EX-527 (a SIRT1 inhibitor (Cat. No. E7144, Sigma Aldrich, MA, USA) or 1 µM Exendin (9-39) amide (a GLP1 receptor antagonist (Cat. No. ab141101, Abcam, Cambridge, UK). The doses of EX-527 and Exendin (9-39) amide were pre-determined in our preliminary data, where they completely inhibited SIRT1 activity and cAMP generation, respectively, by more than 90%. All drugs were always prepared in DMSO and diluted with PBS (pH = 7.4). In all experiments, the DMSO final concentration was always 0.05%.
Determination of cell viability
The viability of cells of all treatments was determined using a colorimetric kit (cell counting kit-8/CCK-8) (Cat No. CK04-13, Dojindo, Japan). Briefly, part of the culture medium (100 μL) was discarded and replaced with an equal volume of fresh medium containing 10% of CCK8 solution. The plate containing the cultured cells was incubated at 37°C for 3 h, and the absorbance was read at 450 nm. Cell viability was calculated as a percent using the following formula ([Abs of the sample−Abs of the blank]/[Abs of the control−Abs of the blank]). The assay followed the manufacturer’s instructions.
Determination of LDH activity in the supernatants
The cells of all treatments were centrifuged for 5 min at 200 x g at 4°C. The pellets were discarded, and the resulting supernatants were collected for analysis of LDH using an assay kit (Cat. No. ab102526; Abcam, Cambridge, UK), which is based on the LDH-dependent reduction of NAD+ to NADPH which can be detected using a special probe at 450 nm. In brief, 50 µL of the prepared reaction mixture was added to 20 µL the pre-adjusted, tested sample, and the color was read immediately at 450 nm. The activity of LDH was calculated from a standard curve.
Determination of apoptosis (ssDNA assay)
The levels of cell apoptosis in all treated groups were calculated using the ssDNA assay ELISA kit (Cat No. APT225, Millipore, USA). All steps followed the manufacturer’s instructions. In the beginning, cells were pelleted (200 x g, 5 min, 4°C) and fixed for 30 min at room temperature with 200 µL of the provided fixative. Then, the fixative was removed followed by the addition of 100 µL formamide solution. The plate was incubated at room temperature for 10 min and then heated in an oven for another 10 min at 75°C to denature DNA. After that, the plate was allowed to cool in the fridge, and the formamide solution was discarded. After that, all wells were blocked by the addition of 3% skim milk (prepared in distilled water) and incubated at 37°C for another 1 h. Then, the milk was discarded, and the cells of all wells were incubated with 100 μL of the provided mouse monoclonal anti-ssDNA antibody. The plate was incubated again at room temperature for an extra 30 min. All wells were then washed (3X/2 min) with 250 µL of the provided washing. Following, the membranes were incubated at room temperature with the and HRP-secondary anti-mouse secondary antibody (200 μL) for 30 min followed by the addition of 100 μL ABTS solution. Finally, 100 µL of the supplied stop solution was added to each well and the absorbance of all wells was read at 405 nm.
Rates of cell migration and invasiveness of both HT29 and HCT1165 were evaluated with or without Exendin-4 treatment as described in our laboratories previously (El-Kott et al., 2019). To evaluate the cell migration, 5 × 104 cells (100 µL) and 200 μL serum-free medium (SFM) were plated on the top of the 8-μm pore size Corning Transwell chambers (diameter of 6.5 mm) (USA) and either treated Exendin-4 (45 µM for HT29 and 35 µM for HCT1165) in the presence or absence of the inhibitors (i.e., 10 µM EX-527 or 1 µM Exendin (9-39) amide). Control cells were incubated with 0.05% DMSO. In all treatments, 500 µL 10% FBS was added to the bottom of the chamber. The cells were incubated in this setting for 24 h. Then, the cells presented in the lower chamber were fixed at room temperature with methanol for 10 min. To visualize the cells, they were stained with 0.2% crystal violet and left for 10 min at 25°C and then counted under 10X of a light microscope. A similar procedure, but with 5-mm pore size Corning Transwell chambers that is percolated with 50 Matrigel µL (Cat. No. DLW354263, Sigma Aldrich, USA) was used for the invasion test. However, cells first starved in the SFM for 3 h and then loaded onto the upper chamber. Cell count was done as described in the migration test.
Cell fractionation and homogenates preparation
The cytoplasmic/nuclear fractions of all treated cells were prepared using a commercially available isolation kit (Cat No. 78835 and ThermoFisher Scientific). To prepare total cell homogenates for the western blotting protocol, the cell pellets were homogenized in radioimmunoprecipitation (RIPA) buffer (250 µL), centrifuged at 11000 x g for 10 min at 4°C to isolate the supernatants. Protein levels in the cytoplasmic, nuclear, and total fractions were evaluated using a Bradford assay (Cat. No. 23300, ThermoFisher Scientific, MA, USA). The levels of target protein expression were calculated relative to the expression of β-actin.
Determination of SIRT1 and NF-κB p65 in the nuclear fractions
SIRT1 deacetylase activity and the nuclear activity of NF-κB p65 were determined using assay kits (Cat. No. CY-1151V, Nagano, Japan and TransAM, Cat. No. 40596, Active Motif, Tokyo, Japan) as per the manufacturers’ instructions.
This procedure was done to detect the acetylated protein levels of both β-catenin and NF-κB p65 by western blotting (Cat. No. 206996, Abcam, Cambridge, UK). In brief, 4 µg of the antibodies against β-catenin or NF-κB p65 or normal rabbit IgG antibodies were added to each sample (containing 100 µg proteins), and the total volume was adjusted to 500 µL using the lysis buffer containing the protease inhibitor cocktail. The whole mixture was kept in rotation for 4 h at 4°C. Four different samples, then 30 µL of protein A/G Sepharose beads were added to every reaction and incubated, and the mixture was further incubated for 1 h at 4°C. All reactions were then centrifuged (2000 x g, 3 min) to collect the protein A/G Sepharose beads. These were then washed (3x) with the provided washing buffer. The beads were allowed to dry at room temperature for 20 min and then were loaded with 40 µL 2x Laemmli buffer, boiled for 5 min and then stored at −80°C for western blotting protocol.
Western blotting was conducted in accordance with the methods established by others (Shan et al., 2020; Su et al., 2020). Equal protein concentrations of the all-cell fractions (40 µg/well) were separated using an SDS–polyacrylamide gel electrophoresis (PAGE) for 1 h at 100 V. The bands in every gel were then transferred onto nitrocellulose membranes for another 1.5 h at 80 V. Membranes were then blocked by 5% skimmed milk prepared in Tris-buffered saline-tween-20 (TBST buffer) and blotted using each target antibody (Tab. 1) (prepared in TBST buffer) or with acetylated lysine antibody (Cat. No. 9441, 1:500) for 2 h at room temperature. Membranes were then washed with the TBST buffer (3X/10 min) and then incubated for 1.5 h at room temperature with the HRP-2nd antibodies. After three times washing with the TBST buffer, the bands were developed using a Pierce ECL kit (ThermoFisher, USA) and scanned and analyzed using the C-Di Git blot scanner (LI-COR, NE, USA).
Data were analyzed on Graph Pad Prism statistical software package (version Differences using the one-way ANOVA and Tukey’s test as post hoc. The level of significance was considered at P < 0.05.
Exendin-4 inhibits cell survival and induces cell death in both HT29 and HCT116 CRC cell lines in a dose-dependent manner
As shown in the Suppl. Figs. S1A–S1D and S2A–S2D, Exendin-4 significantly and in a dose-dependent manner (10–200 µM) reduced cell survival and increased the release of LDH and levels of ssDNA in both HT29 and HCT119 CRC cell lines as compared to their controls treated with diluted DMSO. The calculated IC50 levels of Exendin-4 in HT29 and HCT116 CRC cells were 44.19 µM and 35.45 µM, respectively (Suppl. Figs. S1A–S1D). These doses were used in the rest of the experiments.
Exendin-4 induces cell death and inhibits cell survival, migration, and invasions in both HT29 and HCT116 CRC cell lines in SIRT1 and GLP-1R-dependent mechanism
In the same line, treating the CRC cells with Exendin-4 at its IC50 (45 µM was for HT29 and 35 µM for HCT116) significantly reduced cell survival, invasion, and migration and increased the release of LDH and levels of ssDNA in both cell lines as compared to DMSO treated cells (Figs. 1A–1F and 2A–2D). However, these effects were completely reversed and restored to their basal control levels in both cell lines when Exendin-4 treated cells were pre-treated with Ex-527, a selective SIRT1 inhibitor, or with Exendin (9-39) amide, a GLP-1R antagonist (Figs. 1A–1F and 2A–2D).
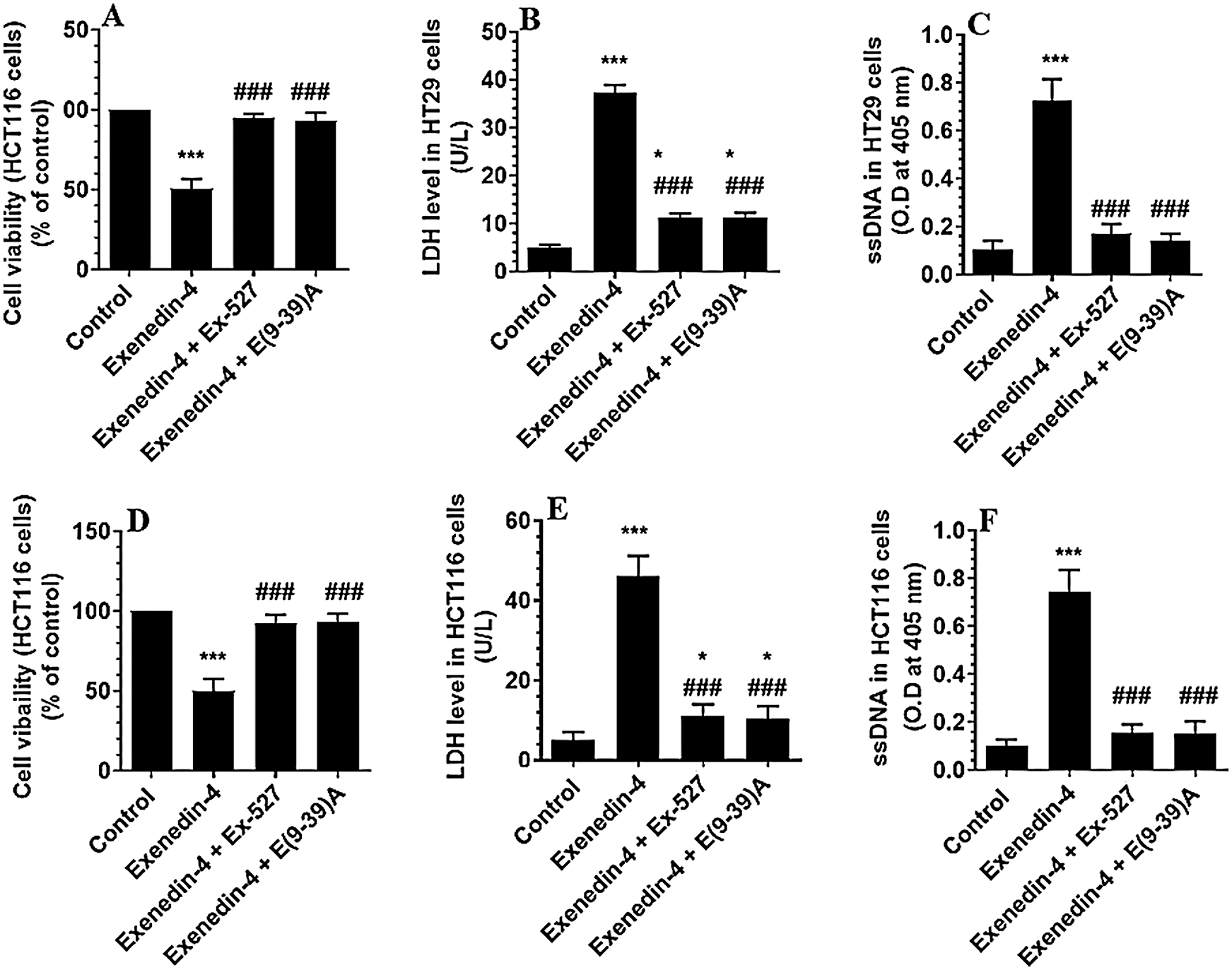
Figure 1: Cell viability and levels of Lactate Dehydrogenase (LDH) and single-stranded DNA (ssDNA) in HT29 (A–C, respectively) and HCT116 (D–F, respectively) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C and 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E (9-39) A) (1 µM), a GLP-1 antagonist. *,***: vs. control (DMSO-treated) at P < 0.05 and P < 0.001, respectively. ###: vs. Exenedin-4-treated cells at P < 0.001.
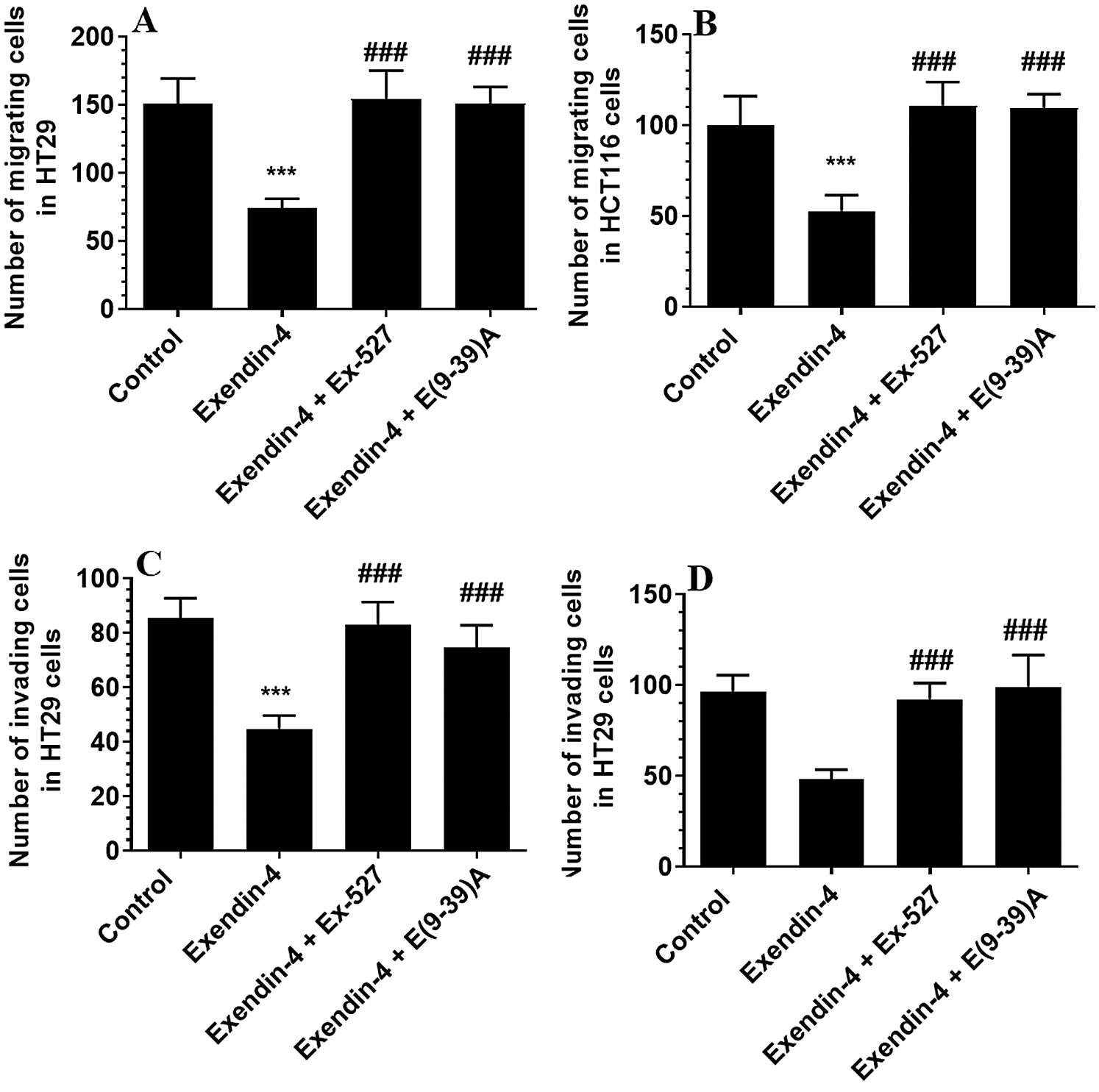
Figure 2: Number of migrating (A–B) and invading cells (C–D) in HT29 and HCT116 colorectal cells, respectively) and their calculated half inhibitory concentration (IC50) (B–D, respectively). The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C and 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E(9-39)A) (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. ***: vs. control (DMSO-treated) at P < 0.001. ###: vs. Exenedin-4-treated cells at P < 0.001.
Exendin-4 has no effects on the expression of GLP-1Rs but significantly stimulates the activity and protein levels of SIRT1 in both HT29 and HCT116 CRC cell lines
The protein levels of GLP-1Rs remained significantly unaltered in both CRC cell types of any treatment (Figs. 3A–3B). However, the nuclear activity of SIRT1 (Suppl. Fig. S3), as well as total protein levels of SIRT1 (Figs. 3C–3D), was significantly increased in Exendin-4-treated cells of both HT29 and HC116 cells as compared to their corresponding control cells treated with DMSO. However, as compared to Exendin-4 treated cells, the activity and protein levels of SIRT1 were significantly reduced to their normal levels in Exendin-4-treated HT29, and HCT116 cells were pre-incubated with either Ex-527 or Exendin (9-39) amide (Suppl. Figs. S3A–S3B and Figs. 3C–3D).
Exendin-4 enhances the activity of GS3Kβ, independent of Akt, in both HT29 and HCT116 CRC cell lines in a SIRT1 and GLP-1Rs-dependent mechanism
As depicted in Figs. 4A and 4B, there was no significant variation in total l protein levels of Akt, as well in the levels of p-Akt (Thr308) between all study groups of both HT29 and HCT116 CRC cell lines as compared to DMSO-treated cells. These data indicate no effect of Exendin-4 on Akt activity in both cell lines. However, the catalytic activity of GS3Kβ is inhibited by phosphorylation at Ser9, whereas its apoptotic activity is increased by Tyr216 phosphorylation (Jain et al., 2017). Herein, the protein level p-GS3K (Ser9) were significantly decreased, whereas protein levels of p-GS3Kβ (Tyr216) were significantly increased in Exendin-4-treated HT29 and HCT116 cells as compared to their control groups (Figs. 5A–5D), thus suggesting activation of the catalytic and apoptotic activity of GS3Kβ. Also, levels of Bax and cleaved caspase-3 were significantly increased in both cell lines which were treated with Exendin-4 as compared to their control untreated cells (Figs. 6A–6D). On the contrary, all these effects afforded by Exendin-4 on the phosphorylation of GS3Kβ and total protein levels of Bax and cleaved caspase-3 were completely reversed in both Exendin-4-treated HT29 and HCT116 cells when they were pre-incubated with Ex-527 or Exendin (9-39) amide (Figs. 5A–5D and 6A–6D).
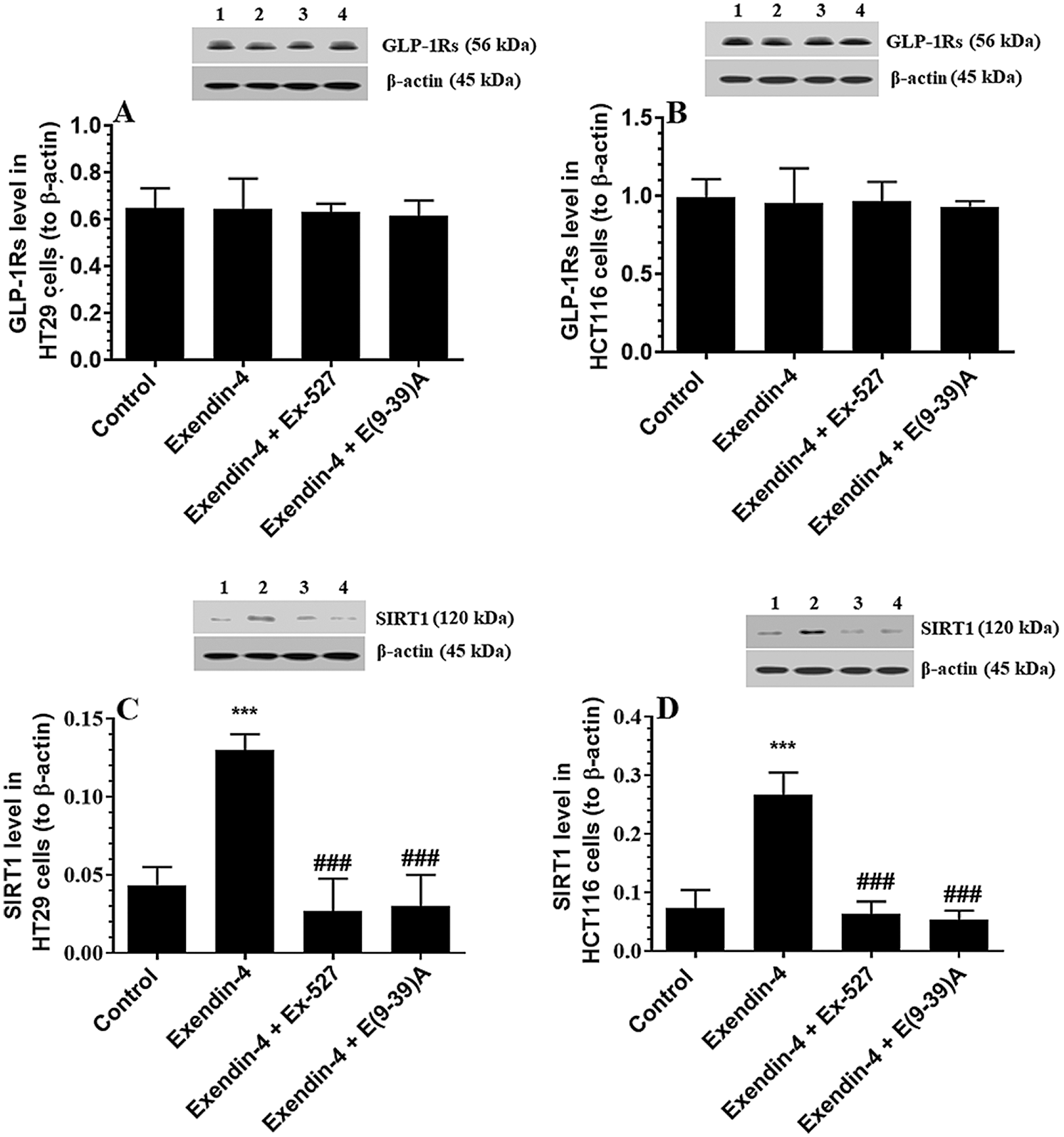
Figure 3: Protein levels of GLP1 and SIRT1 in HT29 (A–C, respectively) and HCT116 (B–D, respectively) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C and 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E(9-39)A) (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. ***: vs. control (DMSO-treated) at P < 0.00. ###: vs. Exenedin-4-treated cells at P < 0.001. Lane 1: control cells; Lane 2: Exendin-4-treated cells; Lane 3; Exendin-4 + Ex-527-treated cells. Lane 4: Exendin-4 + E(9-39)A-treated cells, respectively.
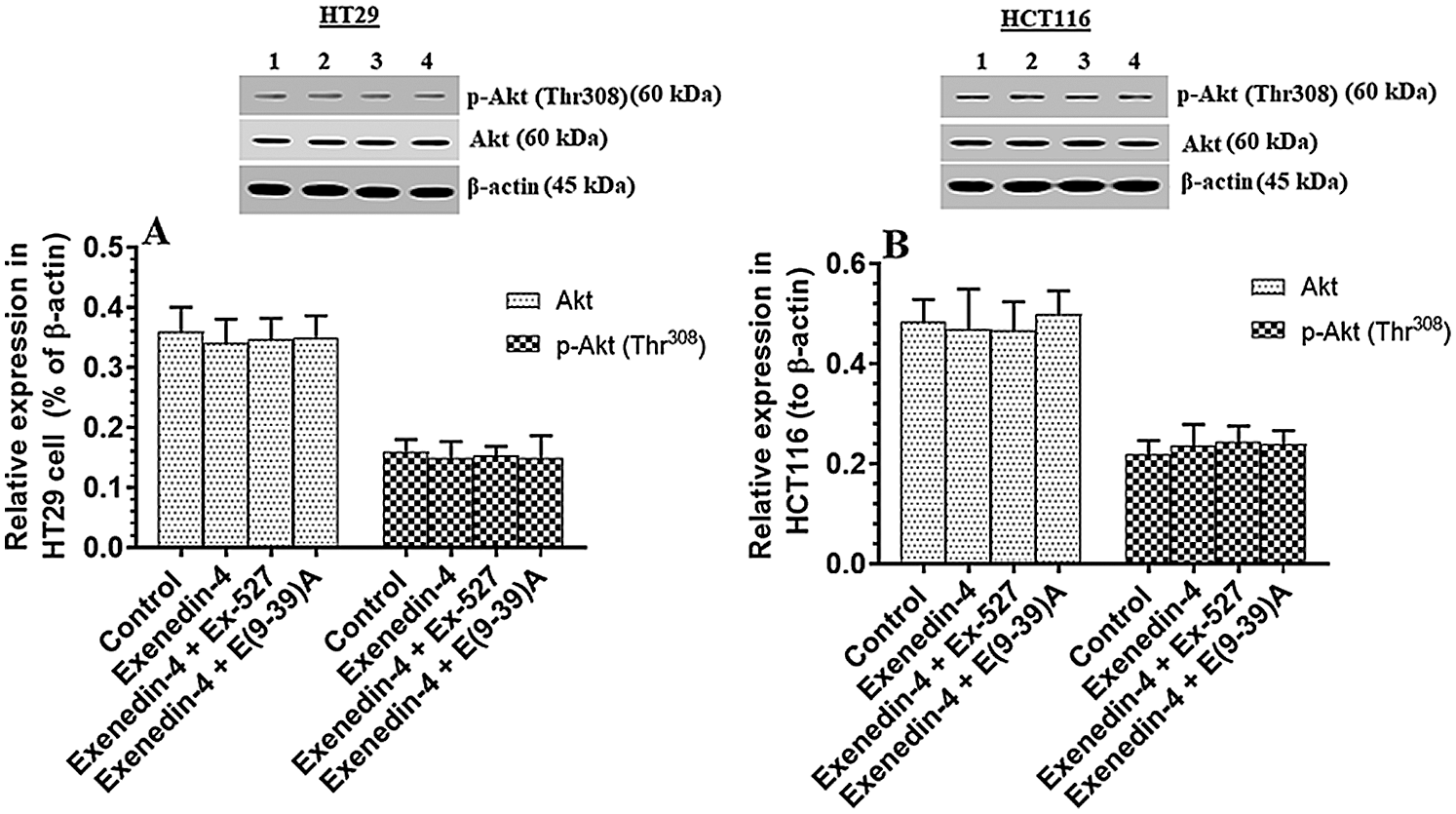
Figure 4: Protein levels of Akt and phosphor-Akt (Thr308) in HT29 (A) and HCT116 (B) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C & 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E(9-39)A) (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. No significant variation in the levels of Akt or p-Akt (Thr308) was detected between all groups of the study of both cell lines. Lane 1: control cells; Lane 2: Exendin-4-treated cells; Lane 3; Exendin-4 + Ex-527-treated cells. Lane 4: Exendin-4 + E(9-39)A-treated cells, respectively. Lane 1: control cells; Lane 2: Exendin-4-treated cells; Lane 3; Exendin-4 + Ex-527-treated cells. Lane 4: Exendin-4 + E(9-39)A-treated cells, respectively.
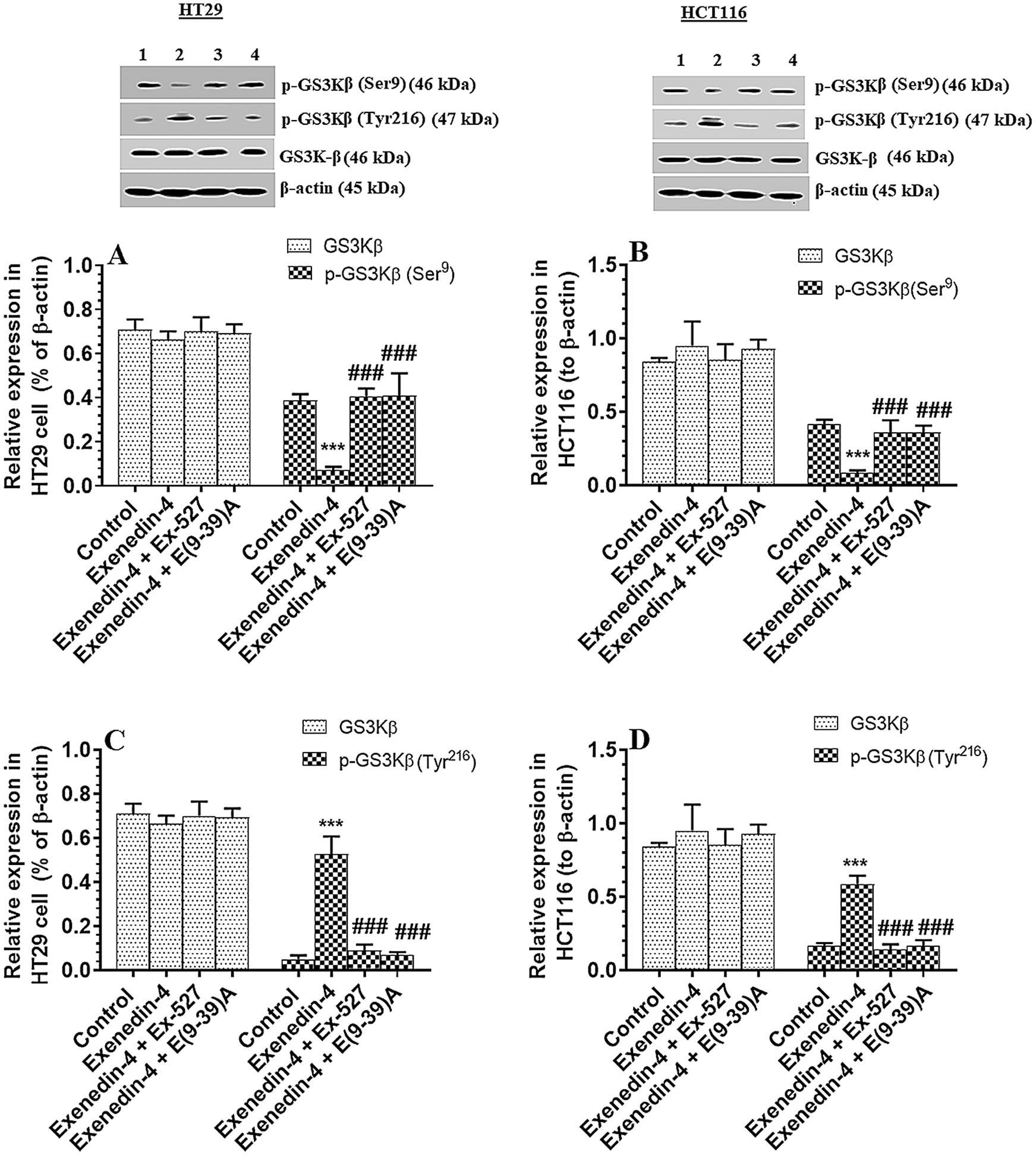
Figure 5: Protein levels of total and p-GS3Kβ (Ser9 and Tyr612) in HT29 (A&C, respectively) and HCT116 (B&D, respectively) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C & 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E(9-39)A) (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. ***: vs. control (DMSO-treated) at P < 0.00. ###: vs. Exenedin-4-treated cells at P < 0.001. Lane 1: control cells; Lane 2: Exendin-4-treated cells; Lane 3; Exendin-4 + Ex-527-treated cells. Lane 4: Exendin-4 + E(9-39)A-treated cells, respectively.
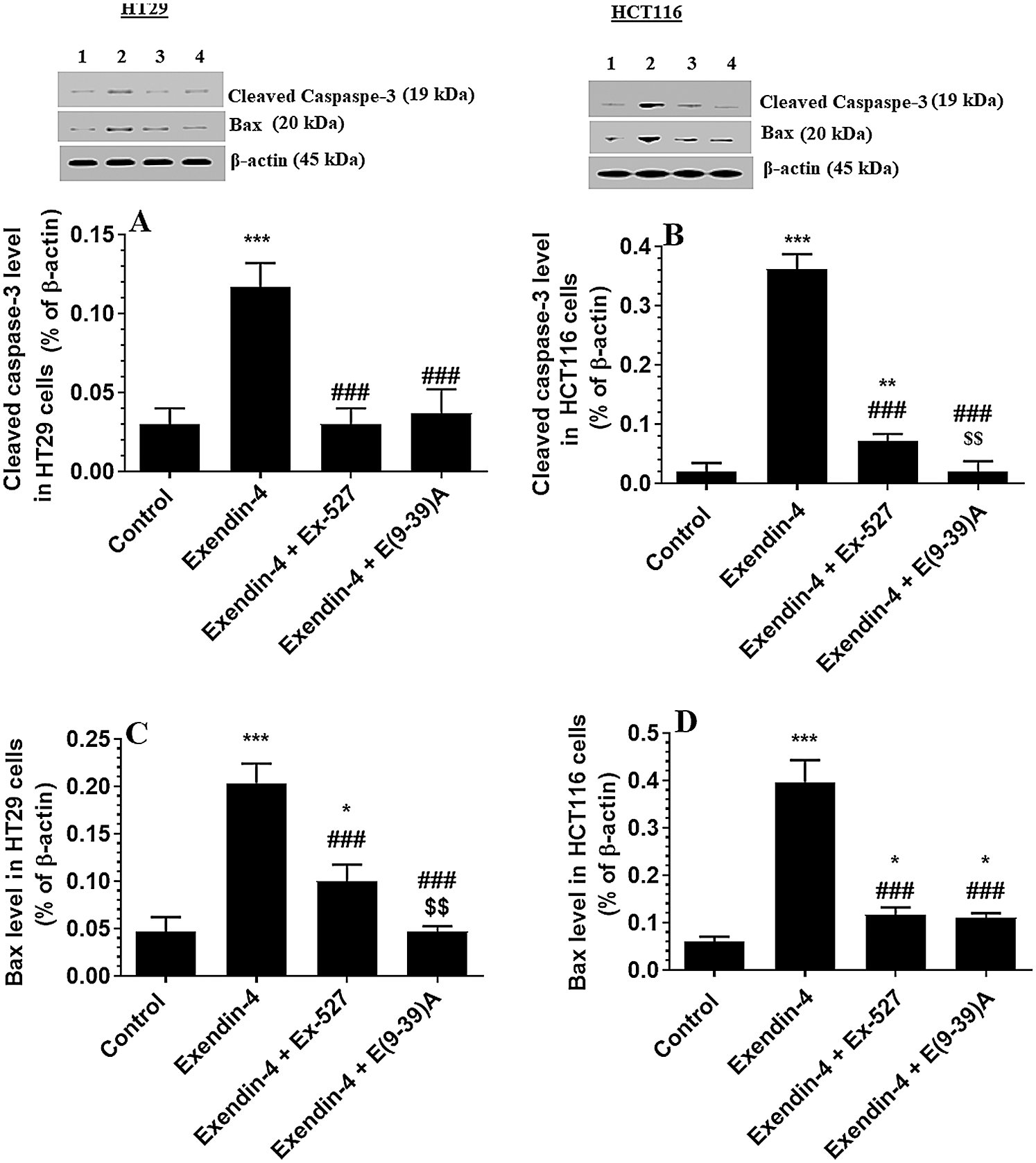
Figure 6: Protein levels of Cleaved caspaspe-3 and Bax in HT29 (A&C, respectively) and HCT116 (B&D, respectively) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C & 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E(9-39)A) (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. *,**,***: vs. control (DMSO-treated) at P < 0.05; 0.01, and 0.001, respectively. ###: vs. Exenedin-4-treated cells at P < 0.001. $$$: vs. Exendin-4 + Ex527 at P < 0.001. Lane 1: control cells; Lane 2: Exendin-4-treated cells; Lane 3; Exendin-4 + Ex-527-treated cells. Lane 4: Exendin-4 + E(9-39)A-treated cells, respectively.
Exendin-4 suppressed total protein level of β-catenin, Acetylated β-catenin, and p-β-catenin (Ser33/37/Thr41) in both HT29 and HCT116 CRC cell lines in a SIRT1 and GLP-1Rs-dependent mechanism
GS3Kβ phosphorylates β-catenin at Ser33/37 and Thr41 to enhance its ubiquitination and proteasome degradation (Rajkumar, 2016). Also, SIRT1 inhibits β-catenin activity by deacetylation at Lys345 (Levy et al., 2004; Wolf et al., 2002). Associated with the increased activity of GS3Kβ and SIRT1 and levels of SIRT, Exendin-4 significantly decreased total and acetylated protein levels of β-catenin and increased protein levels of p-β-catenin (Ser33/37/Thr41) in both cell lines as compared to control cells treated with DMSO (Figs. 7A–7D). Also, it reduced levels of Axin-2, a major downstream target of β-catenin in both HT29 and HCT166 CRC cells (Figs. 8A and 8B). These data suggest that Exendin-4 inhibits β-catenin signaling in both HT29 and HCT116 CRC cells. On the other hand, total and acetylated protein levels of β-catenin, as well as levels of p-β-catenin (Ser33/37/Thr41), were not significantly different when Exendin-4 + Ex-527 and Exendin-4 + Exendin (9-39) amide-treated cells of both cell lines were compared to their control groups (Figs. 7A–7D and 8A–8B), thus suggesting that the suppressive effect of Exendin-4 on β-catenin activation is SIRT1 and GLP-1R-dependent.
Exendin-4 inhibits the activation of NF-κB in both HT29 and HCT116 CRC cell lines in a SIRT1 and GLP-1Rs-dependent mechanism
The activity of NF-κB p65 is negatively controlled by GS3Kβ (phosphorylation at Ser468) and by deacetylation by SIRT1 (Buss et al., 2004; Yang et al., 2012). The activity of NF-κB was significantly decreased in both cell lines which were treated with Exendin4 as compared to their control cells (Suppl. Figs. S3C–S3D). In addition, the protein levels of p-NFκB p65 (Ser468) were significantly increased, and its acetylated levels were significantly decreased in both HT29 and HCT116 cells treated with Exendin-4 (Figs. 9A–9B). However, there was no significant variation in total protein levels in all groups of treatments for both cell lines (Figs. 9A–9B). All these effects were completed prevented in both cells, which were treated with Exendin-4 but pre-treated with Ex-527 or E (9-39) A (Suppl. Figs. S3C–S3D; Figs. 9A–9D).
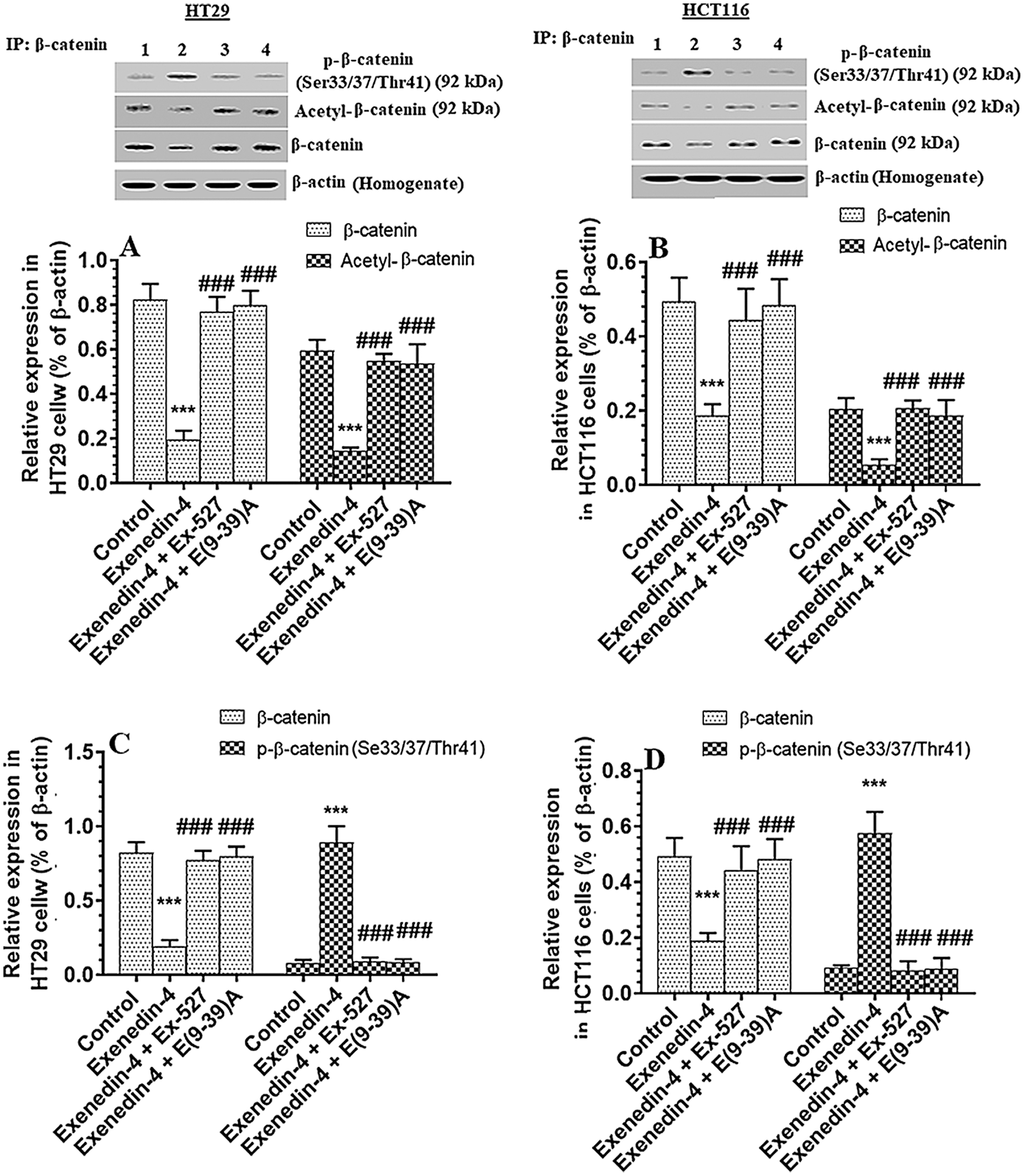
Figure 7: Protein levels of total, p-(Ser33/37/Thr41), and acetylated β-catenin in HT29 (A&C, respectively) and HCT116 (B&D, respectively) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C & 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E(9-39)A) (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. ***: vs. control (DMSO-treated) at P < 0.001, respectively. ###: vs. Exenedin-4-treated cells at P < 0.001. Lane 1: control cells; Lane 2: Exendin-4-treated cells; Lane 3; Exendin-4 + Ex-527-treated cells. Lane 4: Exendin-4 + E(9-39) A-treated cells, respectively.
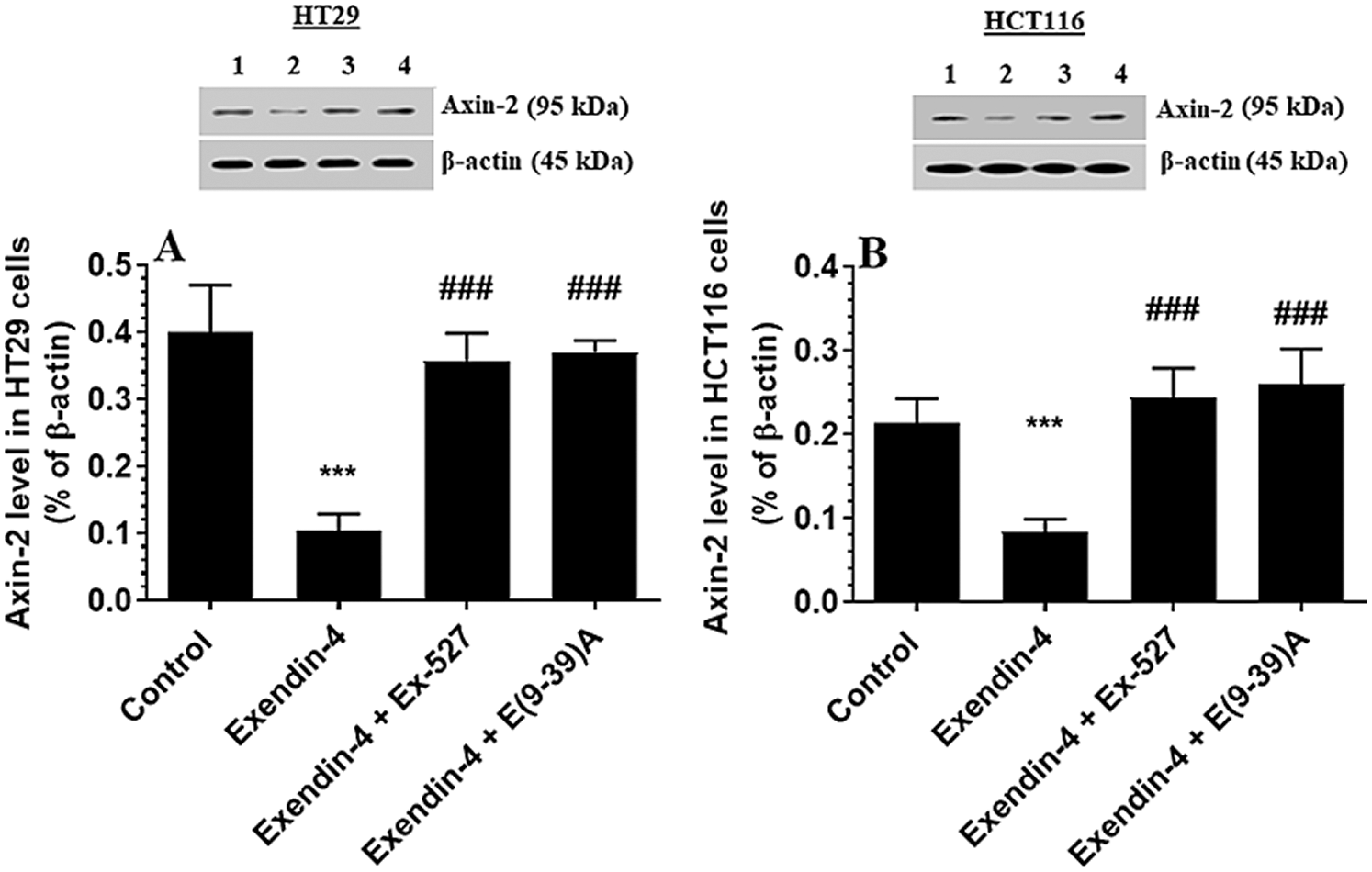
Figure 8: Protein levels Axin2 in HT29 (A) and HCT116 (B) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C & 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E(9-39)A) (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. ***: vs. control (DMSO-treated) at P < 0.001, respectively. ###: vs. Exenedin-4-treated cells at P < 0.001. Lane 1: control cells; Lane 2: Exendin-4-treated cells; Lane 3; Exendin-4 + Ex-527-treated cells. Lane 4: Exendin-4 + E(9-39)A-treated cells, respectively.
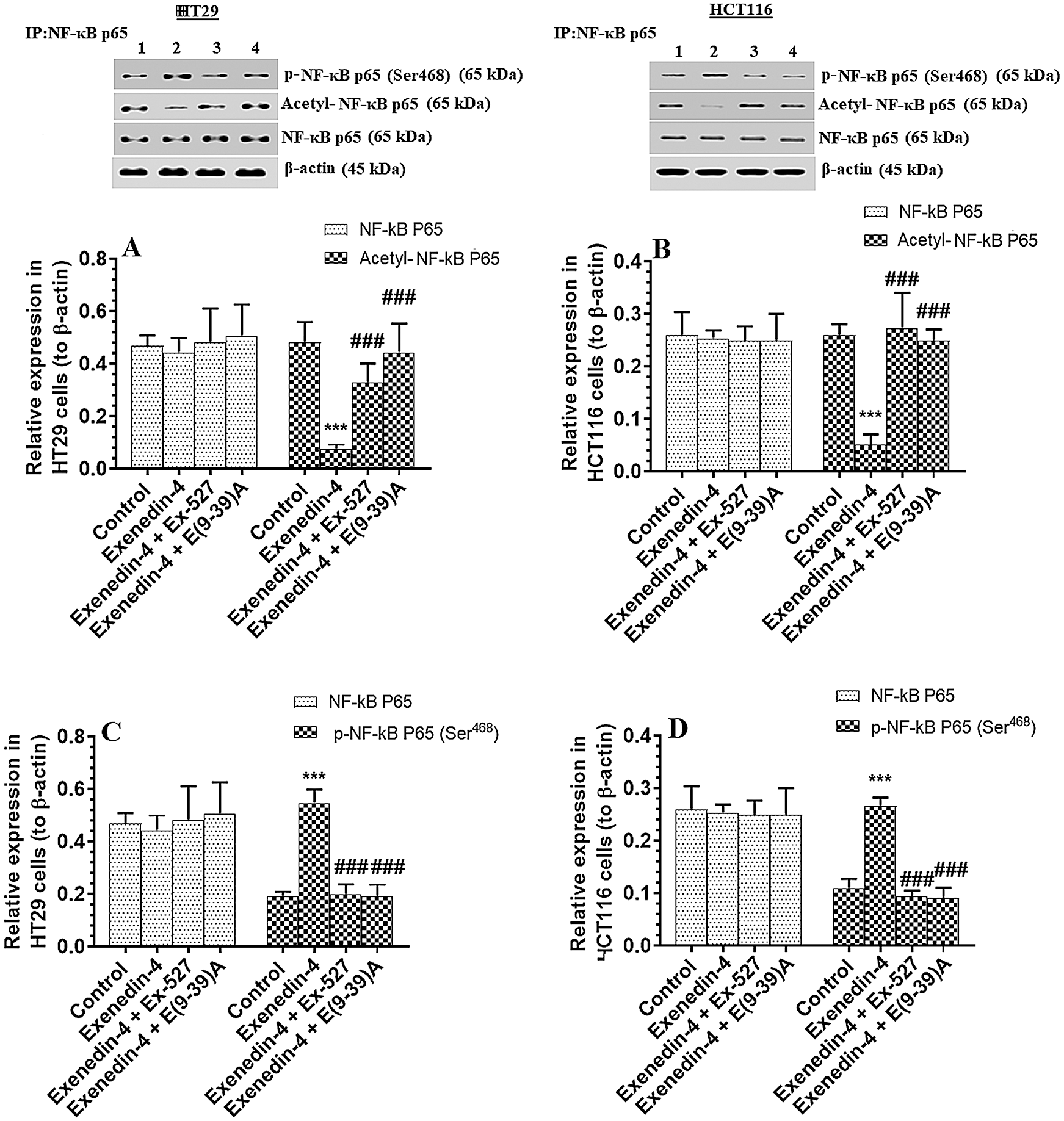
Figure 9: Protein levels of total, p-NF-κB p65 (Ser468) and acetylated NF-κB p65 in HT29 (A&C, respectively) and HCT116 (B&D, respectively) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C & 5% CO2) with or without 1 h pre-incubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (E(9-39)A) (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. ***: vs. control (DMSO-treated) at P < 0.00. ###: vs. Exenedin-4-treated cells at P < 0.001. Lane 1: control cells; Lane 2: Exendin-4-treated cells; Lane 3; Exendin-4 + Ex-527-treated cells. Lane 4: Exendin-4 + E(9-39)A-treated cells, respectively.
The salient findings of this study show that Exendin-4 inhibited the survival and invasiveness of two colorectal cancer (CRC) cell lines, HT29 and HCT116, by suppressing β-catenin activation. These effects were associated with increasing the activity GS3Kβ, inhibiting NF-Kβ activity, and upregulating and activating SIRT1. Further investigations have revealed that the anti-tumorigenesis effect of Exendin-4 is GLP-1 and SIRT1-dependent as pre-incubating the cells with Exendin-4 (9-39) amid, a GLP-1 antagonist or EX-527, a selective SIRT1 inhibitor, completely prevented all the effects afforded by Exendin-4. A summary of the protective effect of these pathways is shown in graphical abstract.
Although the tumor suppressor and the inhibitory effect of Exendin-4 have been confirmed in many solid tumors, few studies have examined its role in a CRC, and contradictory results exist. Koehler et al. (2011) have shown that Exendin-4 induced cell apoptosis and reduced colony formation in murine CT26cells line by increasing intracellular levels of cyclic adenosine monophosphate (cAMP) and inhibiting GS3Kβ and ERK1/2 signaling (Koehler et al., 2011). On the opposite, He et al. (2017) have shown the Exendin-4 does not modify the growth and survival of human colon cancer cells, including Colo320, Caco2, SW480, and LOVO. This variation was attributed to the presence or absence of GLP-1Rs between these cells where the expression of GLP-1Rs was abundant in the CT26 cell lines but absent in all the above-mentioned tested human cell lines. In our preliminary data, we first tested the expression of GLP-1Rs in several cell lines, including HT29 and HCT116, DLD-1, SW480, CaCo2, and CT26, using real-time PCR and western blotting techniques with a very specific monoclonal antibody. Among all, we have found that GLP-1Rs are abundantly expressed in HT29 and HCT116 and to a less extent in CT26 cells, but completely absent in all other tested cells (data not shown except for HT29 and HCT116). Hence, we have decided to continue our experiments using HT29 and HCT116 cell lines.
While the lack of GLP-1Rs on SW480 and CaCo2 supports the data presented by He et al. (2017), our findings contradict their results from the perspective of GLP-1Rs expression in the HT29 cells. This variation could be explained by the age of the cells used, freezing and thawing, culturing conditions, and the specificity of the antibody used. Indeed, several commercial antibodies are not specific to GLP-1Rs (Ussher and Drucker, 2014). While we have used monoclonal specific antibodies, He et al. (2017) have used they have used two polyclonal antibodies. Supporting our data, Saber-Ayad et al. (2018) have shown that both HT29 and HCT116 cell lines express GLP-1Rsinn in abundant quantities. Further evidence shown in this study is the all the anti-tumorigenesis effects exerted by Exednin-4 were significantly attenuated by the use of Exendin (9-39), the antagonist of the GLP-1R.
However, the aberrant activation of the β-catenin is believed to be a major mechanism underlying the development and progression of several solid tumors, including CRC (Bennecib et al., 2000). The activation of this pathway triggers the transcription of several targets involved in cell inflammation, survival, division, proliferation, and migration (Hanahan and Weinberg, 2011; Polakis, 2012). Of interest, genetic silencing or pharmacological inhibition of this cellular molecule afforded potent anti-tumorigenesis effects on Serval cell lines of CRC (Cong et al., 2003; Green et al., 2001; Mologni et al., 2010; Morin et al., 1996; Polakis, 2012; Tetsu and Mccormick, 1999; van de Wetering et al., 2002; Verma et al., 2003). Supporting this evidence, we are demonstrating here that Exendin-4, and at least, by inhibiting β-catenin activation, can induce cell death and attenuate the proliferation and invasiveness of both T29 and HCT116 cell lines, in vitro. Indeed, Exendin-4 at an IC50 dose of 36.6 µM increased the phosphorylation of β-catenin (Ser33/Ser37/Thr41), which are the main site for the action of the GS3Kβ (Metcalfe and Bienz, 2011; Rajkumar, 2016). Besides, it significantly reduced protein levels of Axin2, which is considered a major marker of β-catenin activation (Lustig et al., 2002; Mccubrey et al., 2014; Rajkumar, 2016). Of note, the inhibition of this β-catenin is mediated by a cytoplasmic proteome degradation due to phosphorylation GS3Kβ or by deacetylation by SIRT1 (Cho et al., 2012; Lin and Fang, 2013; Metcalfe and Bienz, 2011; Salaroli et al., 2008). In the first scenario, it is well-reported that activation of GS3Kβ is indispensable for cytoplasmic degradation and inhibition of β-catenin (Metcalfe and Bienz, 2011; Rajkumar, 2016). Of note, phosphorylation of the Ser9 residue inactivates GS3Kβ, whereas the phosphorylation of Tyr216 residue activates it (Jacobs et al., 2012). Therefore, we have examined the effect of Exendin-4, in both cell lines, on the activity of GS3Kβ and the level/activity of SIRT1 Interestingly, Exendin-4 significantly suppressed the phosphorylation of GS3Kβ at its Ser9 and concomitantly increased its phosphorylation at Tyr216, thus implicated activation. These effects occurred without altering the activity of Akt, a major upstream target of GS3Kβ. At the same time, it increased the levels and activity of SIRT1. Taken together, these data suggest the anti-tumorigeneses of Exednin-4 is mediated by its ability to inhibit β-catenin, at least by activation of GS3Kβ and SIRT1.
On the other hand, inflammation is a significant hallmark in the majority of cancers, which stimulate tumor proliferation, survival, metastasis, and angiogenesis and reprogramming its energy metabolism toward glycolysis and lipid synthesis (Greten and Grivennikov, 2019). The activity of NF-κB is mainly regulated by phosphorylation (Lin and Fang, 2013). GS3Kβ is known to inhibit NF-κB p65 by direct phosphorylation at its Ser468 residue and by deacetylation by SIRT1 (Buss et al., 2004). Besides, SIRT1 can inhibit NF-κB by deacetylation (Lin and Fang, 2013; Yang et al., 2012). However, NF-κB and activation of β-catenin can positively affect each other (Rajkumar, 2016). Indeed, overexpression of β-catenin upregulated βTrCP, which in turn activates NF-κB by stimulating the degradation of IκB-α (Winston et al., 1999). Also, NF-κB can directly activate β-catenin by either downregulating LZTS2 and/or reducing the activation of GS3Kβ (Cho et al., 2008; Rajkumar, 2016). Also, IKKα upregulated and activated β-catenin signaling by inhibiting the GS3Kβ (Carayol and Wang, 2006). Besides, TNF-α is a potent activator of β-catenin signaling through the inhibition of GS3Kβ (Li et al., 2014; Oguma et al., 2008).
In this study, we have also found that Exendin-4 can ameliorate the inflammatory microenvironment in both cell lines by attenuating the activation of NF-κB. In this regard, Exendin-4 not only reduced the phosphorylation of NF-κB at Ser468 but also decreased its acetylation. These data support many other studies which have shown that Exendin-4 is capable of attenuating inflammation in breast cancer as well as in obesity, myocardial infarction, atherosclerosis, and stroke by inhibiting NF-kβ (Athauda and Foltynie, 2016; Guo et al., 2016; Iwaya et al., 2017; Lee et al., 2012; Ma et al., 2014). Although it could be possible that Exendin-4 suppresses β-catenin by inhibiting cell inflammation inflammatory or vice versa, our data also suggest that Exendin-4-induced inhibition of NF-κB is mediated by direct phosphorylation of GS3Kβ and upregulation of SIRT1. In support, Eid et al. (2020) have recently shown that Exendin-4 attenuated cardiac remodeling after myocardial infarction and protects the heart from the damaging effect post-ischemia/reperfusion by activation/upregulation of SIRT1–induced inhibition of NF-κB (Eid et al., 2020).
Another important observation in this study is that pre-incubating the cells with EX-527, a selective SIRT1 inhibitor, or Exendin (9-39), a GLP-1 antagonist, completely prevented Exednin-4-induced cell death and increased cell proliferation, survival, and migration. Besides, they prevented the inhibitory effects of Exendin-4 on NF-κB and β-catenin and increased the phosphorylation of GS3Kβ. These data confirm that the activation of SIRT1 by Exendin-4 underlies its anti-tumorigenesis activity in both CT29 and HCT116 cell lines, an effect that is mediated through GLP-1Rs.
Generally, SIRT1 has a dual effect on cancer, of either acting as a tumor promoter or a suppressor. This matter is still under debate. In some studies, the tumor-suppressive effect has been suggested to its ability to inhibit NF-κB as evident in this study (Kong et al., 2011; Lin and Fang, 2013; Yeung et al., 2004). On the other hand, SIRT1 can stimulate tumor growth and metastasis by several other mechanisms, including the deacetylation of p53 and FOXO-1, as well as promoting autophagy (Rajkumar, 2016). P53 mediated intrinsic cell apoptosis directly by upregulation of Bax (Chipuk et al., 2004). Upon acetylation by SIRT1, p53 is unable to induce cell apoptosis due to the downregulation of Bax. However, this contradicts our results which have shown increased expression of Bax and cleaved caspase-3 even with higher levels of SIRT1. Of note, besides acetylation, the activation of p53 is mediated by a wide array of post-translational mechanisms, including phosphorylation, ubiquitination, methylation, glycosylation, simulation, and neddylation (Jacobs et al., 2012). A balance between these various mechanisms determines the fate of p53. During inflammation, NF-κB antagonizes cell death by decreasing the transcriptional activity of p53 (Xia et al., 2014). On the contrary, the activation of GS3Kβ entails an apoptotic signal in various cells by inhibiting the proapoptotic transcription factor such as heat chock protein-1 and CREB and boosting the activity of other transcription factors such as p53 (Jacobs et al., 2012). Within this view, GS3Kβ can also bind to the c-terminal of p53 to directly phosphorylate it at multiple serine residues to promote its nuclear translocation and transcription activity (Beurel et al., 2004; Jacobs et al., 2012; Qu et al., 2004; Turenne and Price, 2001; Watcharasit et al., 2003). Also, several studies have shown that the phosphorylation of GS3Kβ at Tyr216 is essential for its apoptotic activity (Jacobs et al., 2012; Koehler et al., 2011; Yang et al., 2012). Therefore, the decrease in the activation of NF-κB and the concomitant activation of GS3Kβ could explain the apoptotic effect of Exendin-4 and the associated increase in Bax and cleaved caspase-3 levels the treated cell lines.
However, despite this interesting finding, we still have some limitations. Most importantly, although our data have shown that Exendin-4 anti-tumorigenic effect is mediated mainly by a SIRT1 mechanism and through GLP-1Rs, by the use of inhibitors, knocking down these targets is indispensable to strengthen these findings. Therefore, more investigations using transgenic animals or gene silencing in vitro could support our findings. Besides, given the complicated positive and negative interactions between GS3Kβ, inflammatory cytokines, NF-κB, and β-catenin, the data of this study are not sufficient to determine the initial mechanism that triggers all the remaining events. Hence, further studies by targeting each of these markers individually are needed to explain these data. Furthermore, more studies to explain the precise mechanism by which Exendin-4 activates SIRT1 and the subsequent selective activation of GS3Kβ are highly recommended. Of note, the protein phosphatase PP2A is the best-known activator of GS3Kβ by removing the phosphorylation at Ser9 residue (Bennecib et al., 2000). On the other hand, the proline-rich tyrosine kinase 2 (PYK2) and the Fyn tyrosine kinase are the most common kinases that phosphorylate GS3Kβ at Tyr216 (Lesort et al., 1999). Besides, the GLP-1Rs are of G-coupled protein receptors (GPCRs) type, which normally responds to the ligand-binding in a biased agonism (i.e., some signaling is activated while others are inhibited, at the same time to regulate cell signaling). G protein and β-arrestin1/2 are major substrates activated after GCPRs activation. β-arrestin-2 is an important inducer of PP2A and an inhibitor of NF-kB (Beaulieu et al., 2005; Lei et al., 2018; Tang et al., 2019). Therefore, further studies targeting β-arrestin may help to explain our data.
Finally, the Cmax of exendin-4 after SC injection (10 µg) is approximately 250 pg/mL (approximately 60 pM). Therefore, the selected dose of Exendin-4 used in this study seems to be higher than the physiological levels in both humans and animals. However, given that the use of EX-527 reversed the apoptotic and anti-tumorigenic of Exendin-4. These data suggest that the observed effects are not due to non-specific toxicity. Although this dose is much lower than the doses used in other studies of different cell cancer types, further studies examining the anti-tumorigenic effect of Exendin-4 with lower physiological doses on these cell lines are needed.
In conclusion, the data between our hands still interesting and shows for the first time that Exendin-4, a common GLP-1 agonist that is used to treat diabetes, could be a potent suppressor of CRC. At molecular levels, Exendin-4 acts in serval interconnected mechanisms, including activation of GS3Kβ and inhibiting β-catenin and NF-κB. However, these effects are mediated by the activation of SIRT1 and through the GLP-1R.
Acknowledgement: The authors extend their appreciation to the deanship of Scientific Research at King Khalid University, Abha, KSA for supporting this work under grant number (R.G.P.2/80/41), and the work was supported by the Taif University Researchers Supporting Project Number (TURSP-2020/99), Taif University, Taif, Saudi Arabia. Also, this work was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Fast-track Research Funding Program.
Author Contribution: The study conception and design: Attalla El-kott, Ayman E. El-Kenawy and Mashael Mohammed Bin-Meferij; data collection: Eman R. Elbealy and Ali S. Alshehri; analysis and interpretation of results: Heba S. Khalifa, Ehab E. Massoud and Amira M. Alramlawy; draft manuscript preparation: Attalla El-kott, Ayman E. El-Kenawy and Amira M. Alramlawy; All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data used to support the findings of this study are available from the corresponding author upon request.
Funding Statement: This study was supported by the deanship of Scientific Research at King Khalid University, Abha, KSA for supporting this work under grant number (R.G.P.2/80/41), the work was supported by the Taif University Researchers Supporting Project Number (TURSP-2020/99), Taif University, Taif, Saudi Arabia and this work was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Fast-track Research Funding Program.
Conflicts of Interest: The authors have declared no conflict of interest.
Albanese C, Wu K, D’amico M, Jarrett C, Joyce D, Hughes J, Hulit J, Sakamaki T, Fu M, Ben-Ze’ev A, Bromberg JF, Lamberti C, Verma U, Gaynor RB, Byers SW, Pestell RG (2003). IKKα regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Molecular Biology of the Cell 14: 585–599. DOI 10.1091/mbc.02-06-0101. [Google Scholar] [CrossRef]
Athauda D, Foltynie T (2016). The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: Mechanisms of action. Drug Discovery Today 21: 802–818. DOI 10.1016/j.drudis.2016.01.013. [Google Scholar] [CrossRef]
Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG (2005). An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122: 261–273. DOI 10.1016/j.cell.2005.05.012. [Google Scholar] [CrossRef]
Bennecib M, Gong CX, Grundke-Iqbal I, Iqbal K (2000). Role of protein phosphatase-2A and-1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Letters 485: 87–93. DOI 10.1016/S0014-5793(00)02203-1. [Google Scholar] [CrossRef]
Beurel E, Jope RS (2006). The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Progress in Neurobiology 79: 173–189. DOI 10.1016/j.pneurobio.2006.07.006. [Google Scholar] [CrossRef]
Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, Ruiz-Ruiz C, Cadoret A, Capeau J, Desbois-Mouthon C (2004). GSK-3β inhibition by lithium confers resistance to chemotherapy-induced apoptosis through the repression of CD95 (Fas/APO-1) expression. Experimental Cell Research 300: 354–364. DOI 10.1016/j.yexcr.2004.08.001. [Google Scholar] [CrossRef]
Buss H, Dorrie A, Schmitz ML, Frank R, Livingstone M, Resch K, Kracht M (2004). Phosphorylation of serine 468 by GSK-3β negatively regulates basal p65 NF-κB activity. Journal of Biological Chemistry 279: 49571–49574. DOI 10.1074/jbc.C400442200. [Google Scholar] [CrossRef]
Carayol N, Wang CY (2006). IKKα stabilizes cytosolic β-catenin by inhibiting both canonical and non-canonical degradation pathways. Cellular Signalling 18: 1941–1946. DOI 10.1016/j.cellsig.2006.02.014. [Google Scholar] [CrossRef]
Cho HH, Joo HJ, Song JS, Bae YC, Jung JS (2008). Crossregulation of β-catenin/Tcf pathway by NF-κB is mediated by lzts2 in human adipose tissue-derived mesenchymal stem cells. Biochimica et Biophysica Acta 1783: 419–428. DOI 10.1016/j.bbamcr.2007.08.005. [Google Scholar] [CrossRef]
Cong F, Zhang J, Pao W, Zhou P, Varmus H (2003). A protein knockdown strategy to study the function of β-catenin in tumorigenesis. BMC Molecular Biology 4: 10. DOI 10.1186/1471-2199-4-10. [Google Scholar] [CrossRef]
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004). Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014. DOI 10.1126/science.1092734. [Google Scholar] [CrossRef]
Cho IR, Koh SS, Malilas W, Srisuttee R, Moon J, Choi YW, Horio Y, Oh S, Chung YH (2012). SIRT1 inhibits proliferation of pancreatic cancer cells expressing pancreatic adenocarcinoma up-regulated factor (PAUFa novel oncogene, by suppression of β-catenin. Biochemical and Biophysical Research Communications 423: 270–275. DOI 10.1016/j.bbrc.2012.05.107. [Google Scholar] [CrossRef]
Defronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD (2005). Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 28: 1092–1100. DOI 10.2337/diacare.28.5.1092. [Google Scholar] [CrossRef]
Eid RA, Alharbi SA, El-Kott AF, Eleawa SM, Zaki MSA, El-Sayed F, Eldeen MA, Aldera H, Al-Shudiefat AAS (2020). Exendin-4 ameliorates cardiac remodeling in experimentally induced myocardial infarction in rats by inhibiting PARP1/NF-κB axis in a SIRT1-dependent mechanism. Cardiovascular Toxicology 20: 401–418. DOI 10.1007/s12012-020-09567-5. [Google Scholar] [CrossRef]
El-Kott AF, Al-Kahtani MA, Shati AA (2019). Calycosin induces apoptosis in adenocarcinoma HT29 cells by inducing cytotoxic autophagy mediated by SIRT1/AMPK-induced inhibition of Akt/mTOR. Clinical and Experimental Pharmacology & Physiology 46: 944–954. [Google Scholar]
El-Sahli S, Xie Y, Wang L, Liu S (2019). Wnt signaling in cancer metabolism and immunity. Cancers 11: 904. DOI 10.3390/cancers11070904. [Google Scholar] [CrossRef]
Green DW, Roh H, Pippin JA, Drebin JA (2001). Beta-catenin antisense treatment decreases β-catenin expression and tumor growth rate in colon carcinoma xenografts. Journal of Surgical Research 101: 16–20. DOI 10.1006/jsre.2001.6241. [Google Scholar] [CrossRef]
Greten FR, Grivennikov SI (2019). Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 51: 27–41. DOI 10.1016/j.immuni.2019.06.025. [Google Scholar] [CrossRef]
Guo C, Huang T, Chen A, Chen X, Wang L, Shen F, Gu X (2016). Glucagon-like peptide 1 improves insulin resistance in vitro through anti-inflammation of macrophages. Brazilian Journal of Medical and Biological Research 49: 470. DOI 10.1590/1414-431x20165826. [Google Scholar] [CrossRef]
Hanahan D, Weinberg RA (2011). Hallmarks of cancer: The next generation. Cell 144: 646–674. DOI 10.1016/j.cell.2011.02.013. [Google Scholar] [CrossRef]
He WJ, Yu S, Li WS, Xiao HP (2017). Exendin-4 does not modify growth or apoptosis of human colon cancer cells. Endocrine Research 42: 209–218. [Google Scholar]
Iwaya C, Nomiyama T, Komatsu S, Kawanami T, Tsutsumi Y, Hamaguchi Y, Horikawa T, Yoshinaga Y, Yamashita S, Tanaka T, Terawaki Y, Tanabe M, Nabeshima K, Iwasaki A, Yanase T (2017). Exendin-4, a Glucagonlike Peptide-1 Receptor Agonist, Attenuates Breast Cancer Growth by Inhibiting NF-κB Activation. Endocrinology 158: 4218–4232. DOI 10.1210/en.2017-00461. [Google Scholar] [CrossRef]
Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D (2012). GSK-3β: A bifunctional role in cell death pathways. International Journal of Cell Biology 2012: 930710. [Google Scholar]
Jain S, Ghanghas P, Rana C, Sanyal SN (2017). Role of GSK-3β in regulation of canonical Wnt/β-catenin signaling and PI3-K/Akt oncogenic pathway in colon cancer. Cancer Investigation 35: 473–483. DOI 10.1080/07357907.2017.1337783. [Google Scholar] [CrossRef]
Koehler JA, Kain T, Drucker DJ (2011). Glucagon-like peptide-1 receptor activation inhibits growth and augments apoptosis in murine CT26 colon cancer cells. Endocrinology 152: 3362–3372. DOI 10.1210/en.2011-1201. [Google Scholar] [CrossRef]
Kong S, Kim SJ, Sandal B, Lee SM, Gao B, Zhang DD, Fang D (2011). The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. Journal of Biological Chemistry 286: 16967–16975. DOI 10.1074/jbc.M111.218206. [Google Scholar] [CrossRef]
Krishnamurthy N, Kurzrock R (2018). Targeting the Wnt/β-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treatment Reviews 62: 50–60. DOI 10.1016/j.ctrv.2017.11.002. [Google Scholar] [CrossRef]
Lee YS, Park MS, Choung JS, Kim SS, Oh HH, Choi CS, Ha SY, Kang Y, Kim Y, Jun HS (2012). Glucagon-like peptide-1 inhibits adipose tissue macrophage infiltration and inflammation in an obese mouse model of diabetes. Diabetologia 55: 2456–2468. DOI 10.1007/s00125-012-2592-3. [Google Scholar] [CrossRef]
Lei S, Clydesdale L, Dai A, Cai X, Feng Y, Yang D, Liang YL, Koole C, Zhao P, Coudrat T, Christopoulos A, Wang MW, Wootten D, Sexton PM (2018). Two distinct domains of the glucagon-like peptide-1 receptor control peptide-mediated biased agonism. Journal of Biological Chemistry 293: 9370–9387. DOI 10.1074/jbc.RA118.003278. [Google Scholar] [CrossRef]
Lesort M, Jope RS, Johnson GV (1999). Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3β and Fyn tyrosine kinase. Journal of Neurochemistry 72: 576–584. DOI 10.1046/j.1471-4159.1999.0720576.x. [Google Scholar] [CrossRef]
Levy L, Wei Y, Labalette C, Wu Y, Renard CA, Buendia MA, Neuveut C (2004). Acetylation of β-catenin by p300 regulates β-catenin-Tcf4 interaction. Molecular and Cellular Biology 24: 3404–3414. DOI 10.1128/MCB.24.8.3404-3414.2004. [Google Scholar] [CrossRef]
Li D, Beisswenger C, Herr C, Hellberg J, Han G, Zakharkina T, Voss M, Wiewrodt R, Bohle RM, Menger MD, Schmid RM, Stöckel D, Lenhof HP, Bals R (2014). Myeloid cell RelA/p65 promotes lung cancer proliferation through Wnt/β-catenin signaling in murine and human tumor cells. Oncogene 33: 1239–1248. DOI 10.1038/onc.2013.75. [Google Scholar] [CrossRef]
Lin Z, Fang D (2013). The roles of SIRT1 in cancer. Genes & Cancer 4: 97–104. DOI 10.1177/1947601912475079. [Google Scholar] [CrossRef]
Logan CY, Nusse R (2004). The Wnt signaling pathway in development and disease. Annual Review of Cell and Developmental Biology 20: 781–810. DOI 10.1146/annurev.cellbio.20.010403.113126. [Google Scholar] [CrossRef]
Lonborg J, Vejlstrup N, Kelbaek H, Nepper-Christensen L, Jorgensen E, Helqvist S, Holmvang L, Saunamäki K, Bøtker HE, Kim WY, Clemmensen P, Treiman M, Engstrøm T (2014). Impact of acute hyperglycemia on myocardial infarct size, area at risk, and salvage in patients with STEMI and the association with exenatide treatment: Results from a randomized study. Diabetes 63: 2474–2485. DOI 10.2337/db13-1849. [Google Scholar] [CrossRef]
Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J (2002). Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Molecular and Cellular Biology 22: 1184–1193. DOI 10.1128/MCB.22.4.1184-1193.2002. [Google Scholar] [CrossRef]
Ma GF, Chen S, Yin L, Gao XD, Yao WB (2014). Exendin-4 ameliorates oxidized-LDL-induced inhibition of macrophage migration in vitro via the NF-κB pathway. Acta Pharmacologica Sinica 35: 195–202. DOI 10.1038/aps.2013.128. [Google Scholar] [CrossRef]
Mccubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Cocco L, Cervello M, Martelli AM (2014). Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia 28: 15–33. DOI 10.1038/leu.2013.184. [Google Scholar] [CrossRef]
Metcalfe C, Bienz M (2011). Inhibition of GSK3 by Wnt signalling--two contrasting models. Journal of Cell Science 124: 3537–3544. DOI 10.1242/jcs.091991. [Google Scholar] [CrossRef]
Mologni L, Dekhil H, Ceccon M, Purgante S, Lan C, Cleris L, Magistroni V, Formelli F, Gambacorti-Passerini CB (2010). Colorectal tumors are effectively eradicated by combined inhibition of β-catenin, KRAS, and the oncogenic transcription factor ITF2. Cancer Research 70: 7253–7263. DOI 10.1158/0008-5472.CAN-10-1108. [Google Scholar] [CrossRef]
Morin PJ, Vogelstein B, Kinzler KW (1996). Apoptosis and APC in colorectal tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America 93: 7950–7954. DOI 10.1073/pnas.93.15.7950. [Google Scholar] [CrossRef]
Nomiyama T, Kawanami T, Irie S, Hamaguchi Y, Terawaki Y, Murase K, Tsutsumi Y, Nagaishi R, Tanabe M, Morinaga H, Tanaka T, Mizoguchi M, Nabeshima K, Tanaka M, Yanase T (2014). Exendin-4, a GLP-1 receptor agonist, attenuates prostate cancer growth. Diabetes 63: 3891–3905. DOI 10.2337/db13-1169. [Google Scholar] [CrossRef]
Oguma K, Oshima H, Aoki M, Uchio R, Naka K, Nakamura S, Hirao A, Saya H, Taketo MM, Oshima M (2008). Activated macrophages promote Wnt signalling through tumour necrosis factor-α in gastric tumour cells. EMBO Journal 27: 1671–1681. DOI 10.1038/emboj.2008.105. [Google Scholar] [CrossRef]
Polakis P (2012). Wnt signaling in cancer. Cold Spring Harbor perspectives in biology 4: a008052. DOI 10.1101/cshperspect.a008052. [Google Scholar] [CrossRef]
Qu L, Huang S, Baltzis D, Rivas-Estilla AM, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Yoshimura A, Koromilas AE (2004). Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3β. Genes & Development 18: 261–277. DOI 10.1101/gad.1165804. [Google Scholar] [CrossRef]
Rajkumar V (2016). Myeloma today: Disease definitions and treatment advances. American Journal of Hematology 91: 965. DOI 10.1002/ajh.24392. [Google Scholar] [CrossRef]
Saber-Ayad M, Zaher D, Manzoor S, Omar H (2018). PO-453 Effect of GLP-1 on proliferation and migration in pheochromocytoma and colorectal cancer cells. ESMO Open 3: A199–A200. DOI 10.1136/esmoopen-2018-EACR25.474. [Google Scholar] [CrossRef]
Salaroli R, Di Tomaso T, Ronchi A, Ceccarelli C, Cammelli S, Cappellini A, Martinelli GN, Barbieri E, Giangaspero F, Cenacchi G (2008). Radiobiologic response of medulloblastoma cell lines: Involvement of β-catenin? Journal of Neuro-Oncology 90: 243–251. DOI 10.1007/s11060-008-9659-5. [Google Scholar] [CrossRef]
Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, Rupec RA, Gerhard M, Schmid R, Barker N, Clevers H, Lang R, Neumann J, Kirchner T, Taketo MM, van den Brink GR, Sansom OJ, Arkan MC, Greten FR (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152: 25–38. DOI 10.1016/j.cell.2012.12.012. [Google Scholar] [CrossRef]
Shan B, Ai Z, Zeng S, Song Y, Song J, Zeng Q, Liao Z, Wang T, Huang C, Su D (2020). Gut microbiome-derived lactate promotes to anxiety-like behaviors through GPR81 receptor-mediated lipid metabolism pathway. Psychoneuroendocrinology 117: 104699. DOI 10.1016/j.psyneuen.2020.104699. [Google Scholar] [CrossRef]
Siegel RL, Miller KD, Jemal A (2019). Cancer statistics, CA: A. Cancer Journal for Clinicians 69: 7–34. DOI 10.3322/caac.21551. [Google Scholar] [CrossRef]
Su D, Liao Z, Feng B, Wang T, Shan B, Zeng Q, Song J, Song Y (2020). Pulsatilla chinensis saponins cause liver injury through interfering ceramide/sphingomyelin balance that promotes lipid metabolism dysregulation and apoptosis. Phytomedicine 76: 153265. DOI 10.1016/j.phymed.2020.153265. [Google Scholar] [CrossRef]
Taciak B, Pruszynska I, Kiraga L, Bialasek M, Krol M (2018). Wnt signaling pathway in development and cancer. Journal of Physiology and Pharmacology 69: 185–196. [Google Scholar]
Tang J, Zhou S, Zhou F, Wen X (2019). Inhibitory effect of tanshinone IIA on inflammatory response in rheumatoid arthritis through regulating β-arrestin 2. Experimental and Therapeutic Medicine 17: 3299–3306. [Google Scholar]
Tetsu O, Mccormick F (1999). β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398: 422–426. DOI 10.1038/18884. [Google Scholar] [CrossRef]
Thanikachalam K, Khan G (2019). Colorectal cancer and nutrition. Nutrients 11: 164. DOI 10.3390/nu11010164. [Google Scholar] [CrossRef]
Tsutsumi Y, Nomiyama T, Kawanami T, Hamaguchi Y, Terawaki Y, Tanaka T, Murase K, Motonaga R, Tanabe M, Yanase T (2015). Combined treatment with Exendin-4 and Metformin attenuates prostate cancer growth. PLoS One 10: e0139709. DOI 10.1371/journal.pone.0139709. [Google Scholar] [CrossRef]
Turenne GA, Price BD (2001). Glycogen synthase kinase3 β phosphorylates serine 33 of p53 and activates p53’s transcriptional activity. BMC Cell Biology 2: 12. DOI 10.1186/1471-2121-2-12. [Google Scholar] [CrossRef]
Tzotzas T, Karras SN, Katsiki N (2017). Glucagon-Like Peptide-1 (GLP-1) receptor agonists in the treatment of obese women with polycystic ovary syndrome. Current Vascular Pharmacology 15: 218–229. DOI 10.2174/1570161114666161221115324. [Google Scholar] [CrossRef]
Ussher JR, Drucker DJ (2014). Cardiovascular actions of incretin-based therapies. Circulation Research 114: 1788–1803. DOI 10.1161/CIRCRESAHA.114.301958. [Google Scholar] [CrossRef]
van de Wetering M, Sancho E, Verweij C, De Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H (2002). The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111: 241–250. DOI 10.1016/S0092-8674(02)01014-0. [Google Scholar] [CrossRef]
Verma UN, Surabhi RM, Schmaltieg A, Becerra C, Gaynor RB (2003). Small interfering RNAs directed against β-catenin inhibit the in vitro and in vivo growth of colon cancer cells. Clinical Cancer Research 9: 1291–1300. [Google Scholar]
Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS (2003). Glycogen synthase kinase-3β (GSK3β) binds to and promotes the actions of p53. Journal of Biological Chemistry 278: 48872–48879. DOI 10.1074/jbc.M305870200. [Google Scholar] [CrossRef]
Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW (1999). The SCFβ-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes & Development 13: 270–283. DOI 10.1101/gad.13.3.270. [Google Scholar] [CrossRef]
Wolf D, Rodova M, Miska EA, Calvet JP, Kouzarides T (2002). Acetylation of β-catenin by CREB-binding protein (CBP). Journal of Biological Chemistry 277: 25562–25567. DOI 10.1074/jbc.M201196200. [Google Scholar] [CrossRef]
Xia Y, Shen S, Verma IM (2014). NF-κB, an active player in human cancers. Cancer Immunology Research 2: 823–830. DOI 10.1158/2326-6066.CIR-14-0112. [Google Scholar] [CrossRef]
Yamamoto K, Amako M, Yamamoto Y, Tsuchihara T, Nukada H, Yoshihara Y, Arino H, Fujita M, Uenoyama M, Tachibana S, Nemoto K (2013). Therapeutic effect of exendin-4, a long-acting analogue of glucagon-like peptide-1 receptor agonist, on nerve regeneration after the crush nerve injury. BioMed Research International 2013: 1–7. DOI 10.1155/2013/315848. [Google Scholar] [CrossRef]
Yang L, Zhang J, Yan C, Zhou J, Lin R, Lin Q, Wang W, Zhang K, Yang G, Bian X, Zeng A (2012). SIRT1 regulates CD40 expression induced by TNF-α via NF-kB pathway in endothelial cells. Cellular Physiology and Biochemistry 30: 1287–1298. DOI 10.1159/000343318. [Google Scholar] [CrossRef]
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR et al. (2004). Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO Journal 23: 2369–2380. DOI 10.1038/sj.emboj.7600244. [Google Scholar] [CrossRef]
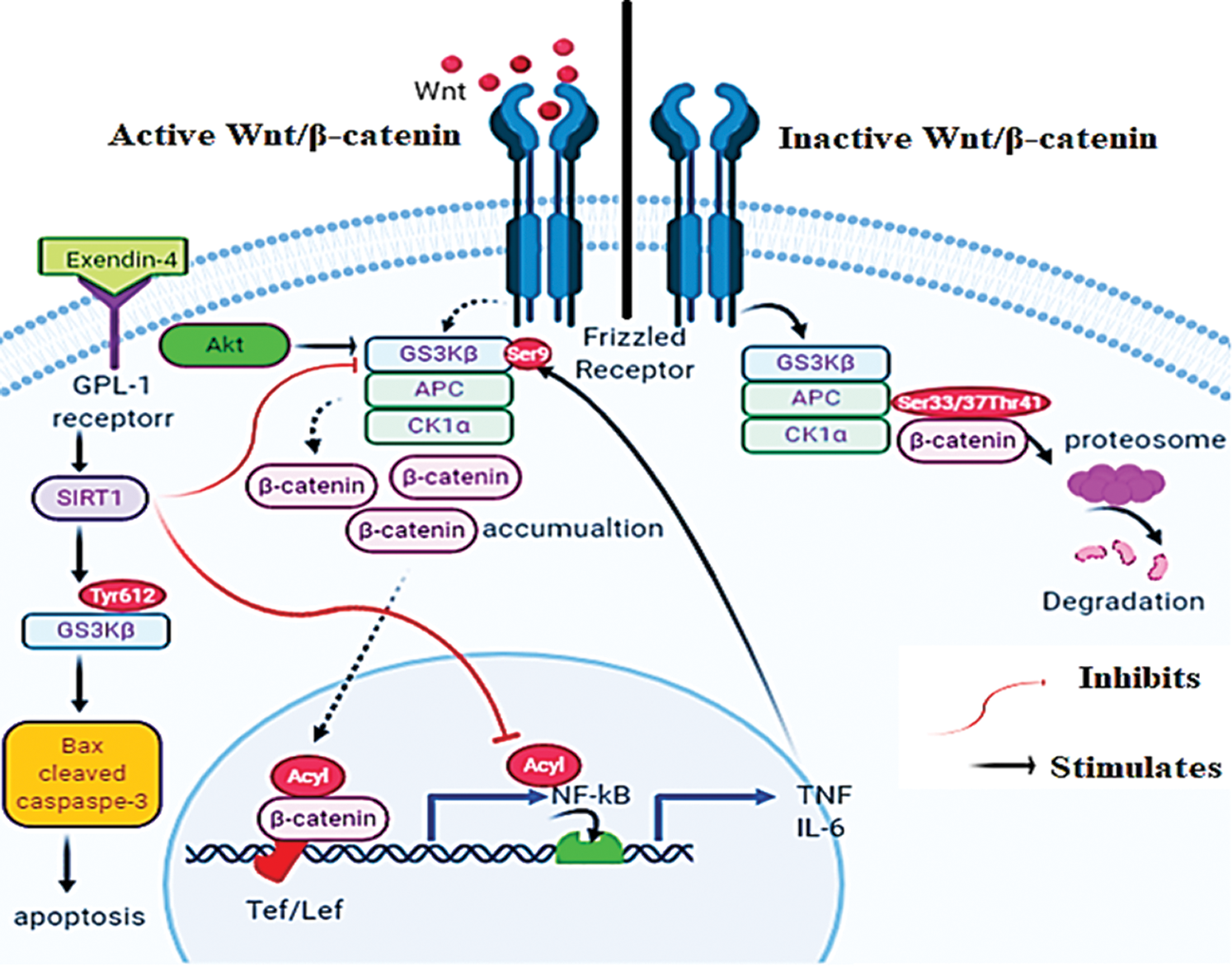
Graphical Abstract
A schematic diagram for presenting the mechanisms by which Exendin-4 induces apoptosis in HT29 and HCT116 colorectal (CRC) cells. In the absence of Wnt, GS3K is active and stimulates the proteasome degradation of β-catenin by phosphorylating it at Ser33/37/Thr41 (Right side). On the other hand, Wnt stimulates the phosphorylation of Dishevelled protein that inhibits the degradation complex by phosphorylating GS3Kβ at Ser9 (Left side). Also, Akt can inhibit GS3Kβ by increasing its phosphorylation at Ser9. These events allow the nuclear translocation of β-catenin that stimulates the transcription activity of NF-κB. Furthermore, the transcriptional activity of NF-κB and β-catenin is activated by acetylation. Exendin-4, and through GLP-1 receptors, increases SIRT1 levels in both HT29 and HCT116 CRC cell lines. In SIRT1-dependent mechanisms (still unknown), Exendin-4 inhibits the phosphorylation of GS3Kβ at Ser9 and thus stimulates β-catenin degradation and decreases its transcriptional activity.
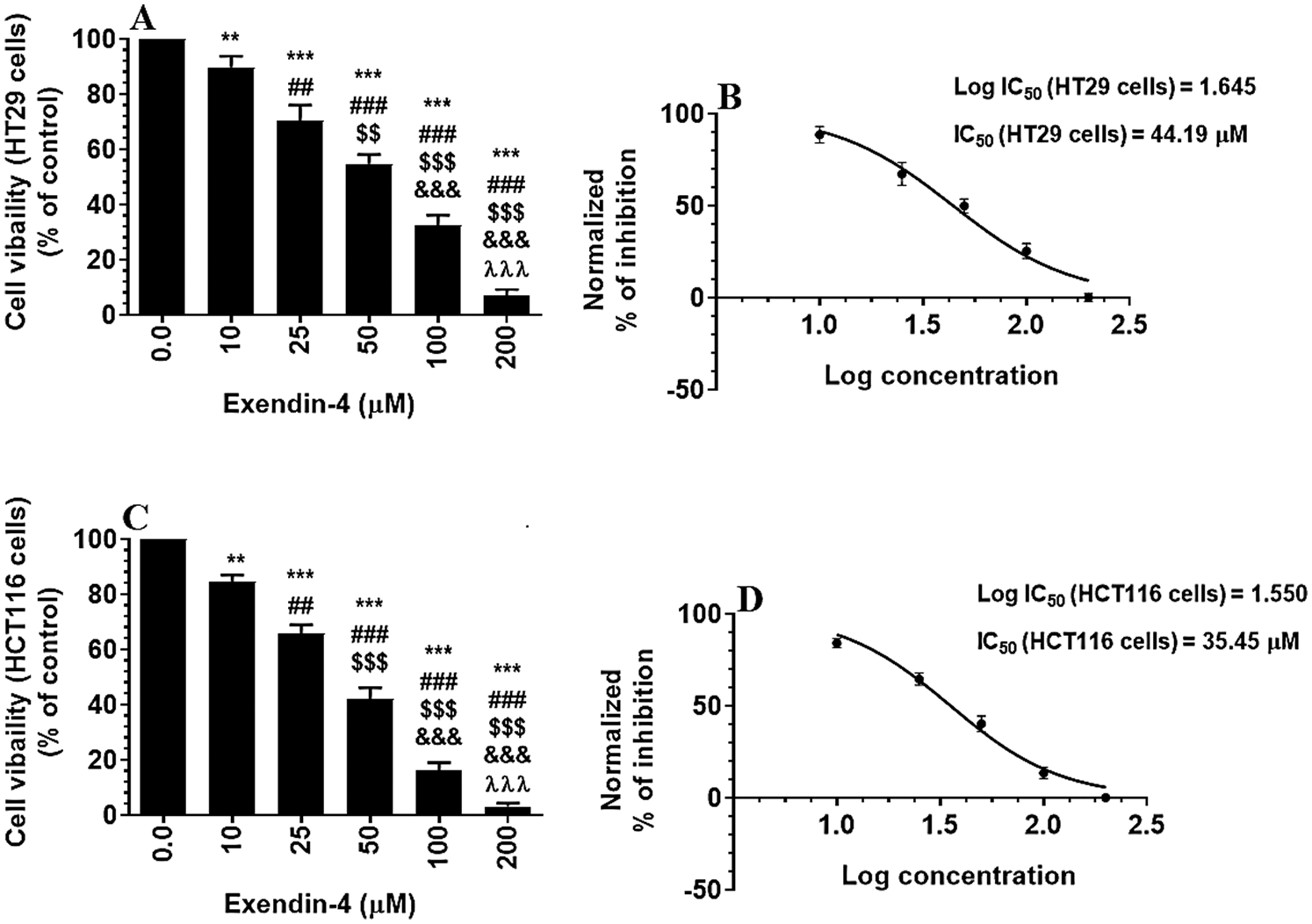
Supplementary Figure S1: Cell Viability in HT29 and HCT116 colorectal cells (A–C, respectively) and their calculated half inhibitory concentration (IC50) (B–D, respectively). The cells were treated with DMSO (0.05%) (control, 0.0 µM of Exendin-4) or with increasing concentration of Exendin-4 (prepared in DMSO) (10–200 µM) for 24 h (37°C and 5% CO2). Data are presented as mean ± SD of three independent experiments, each performed in triplicate. **, ***: vs. control (DMSO-treated) at P < 0.01 and P < 0.001, respectively. ##, ###: vs. 10 µM at P < 0.01 and P < 0.001, respectively. SSS: vs. 25 µM at P < 0.001. &&&: vs. 50 µM at P < 0.001. λλλ: vs. 50 µM at P < 0.001.
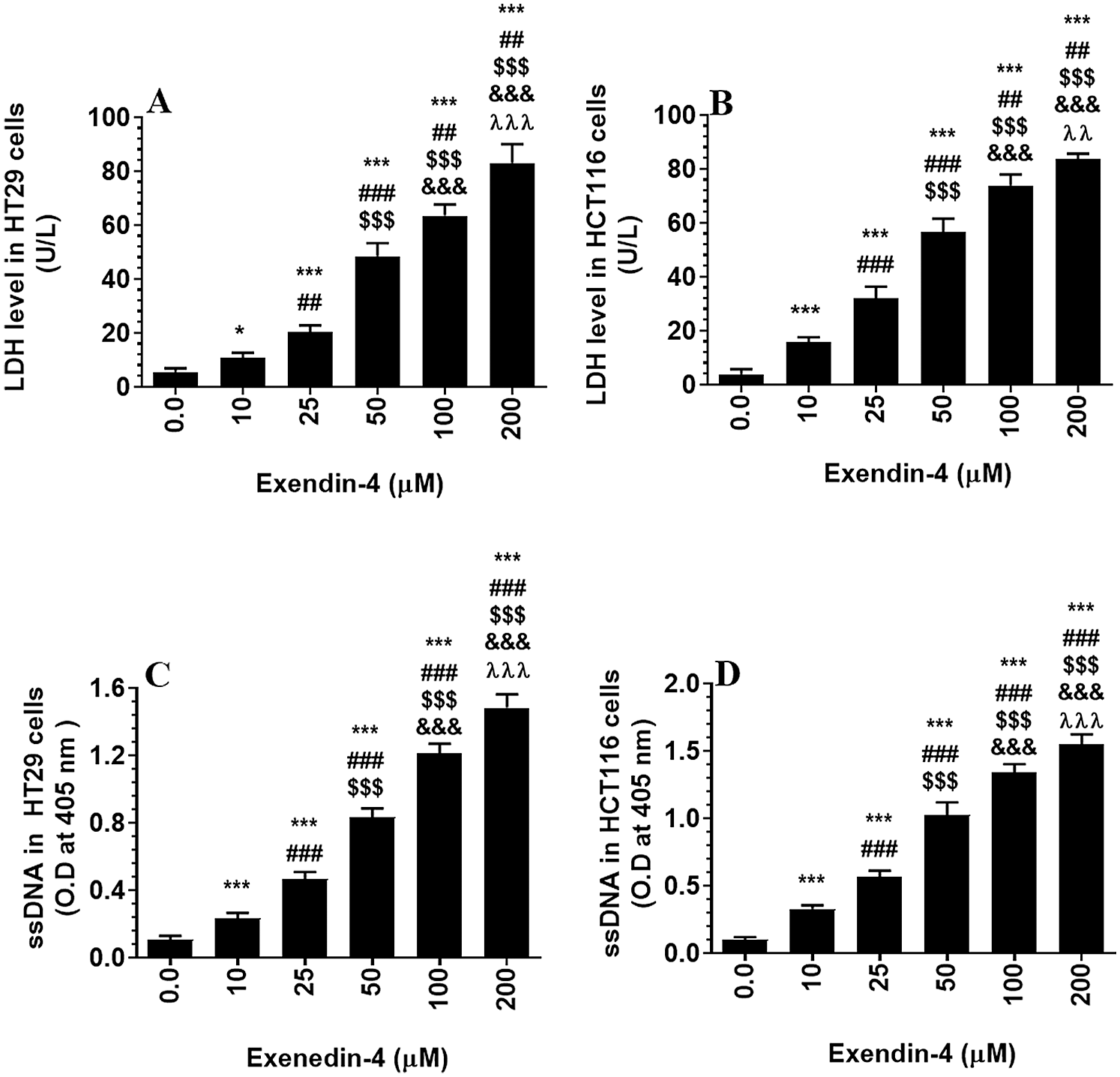
Supplementary Figure S2: Levels of Lactate dehydrogenase (A–B) and single stranded DNA (ssDNA) (B–C) in HT29 and HCT116 colorectal cells, respectively) and their calculated half inhibitory concentration (IC50) (B and D, respectively). The cells were treated with DMSO (0.05%) (control, 0.0 µM of Exendin-4) for 24 h (37°C and 5% CO2) or with increasing concentration of Exendin-4 (prepared in DMSO) (10–200 µM) for 24 h. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. *, ***: vs. control (DMSO-treated) at P < 0.05 and P < 0.001, respectively. ##, ###: vs. 10 µM at P < 0.01 and P < 0.001, respectively. SSS: vs. 25 µM at P < 0.001. &&&: vs. 50 µM at P < 0.001. λλλ: vs. 50 µM at P < 0.001.
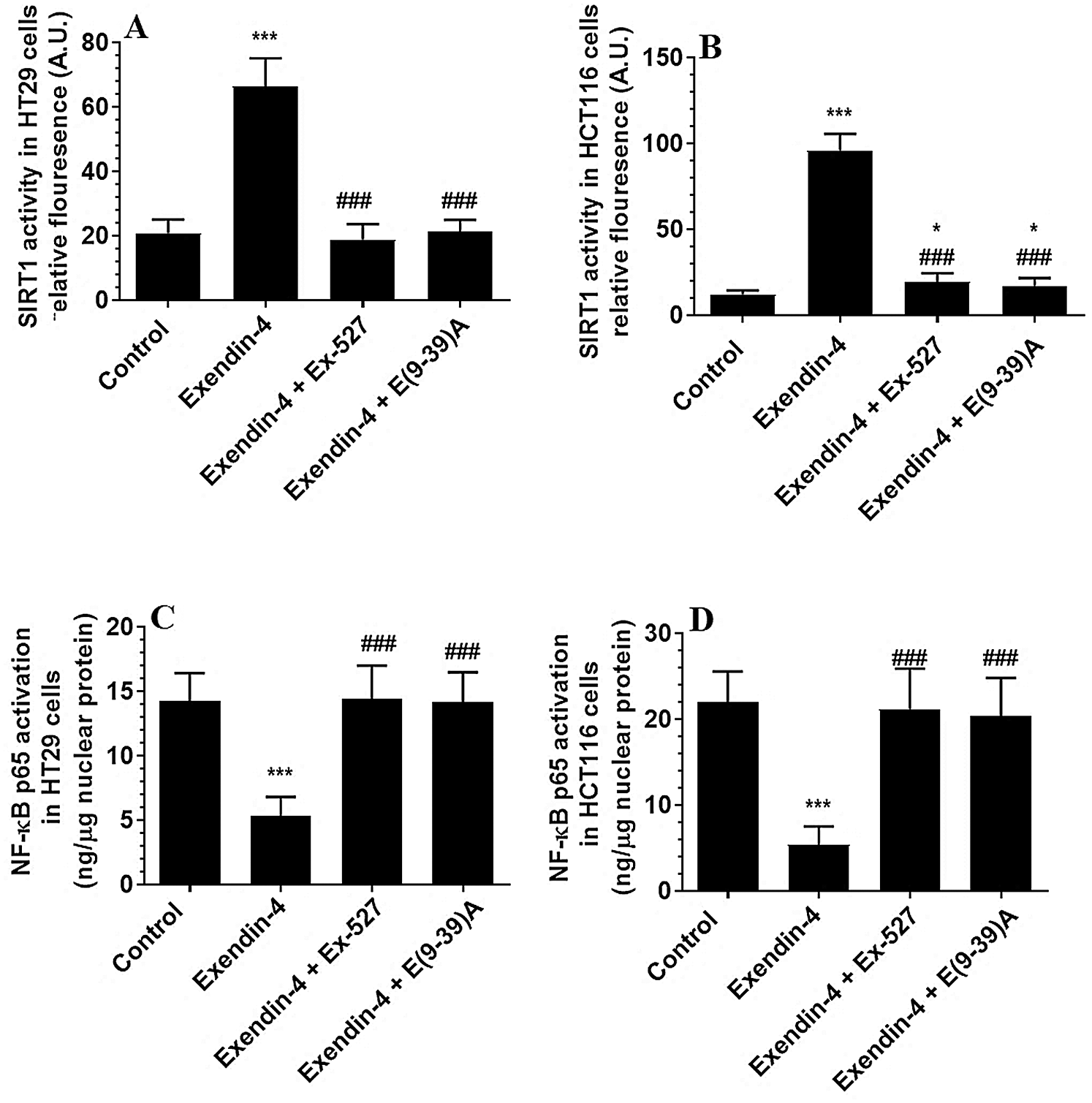
Supplementary Figure S3: The activity of SIRT1 and NF-κB in HT29 (A–B, respectively) and HCT116 (C–D, respectively) colorectal cells of all treated groups. The cells were treated with DMSO (0.05%) (control) in the presence or absence of Exendin-4 at its IC50 (45 µM for HT29 and 35 µM for HCT1165) for 24 h (37°C and 5% CO2) with or without 1 h reintubation with EX-527 (10 µM), A selective SIRT1 inhibitor or Exendin (9-39) amide (1 µM), a GLP-1 antagonist. Data are presented as mean ± SD of three independent experiments, each performed in triplicate. *,***: vs. control (DMSO-treated) at P < 0.05 and P < 0.001, respectively. ###: vs. Exenedin-4-treated cells at P < 0.001.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |