DOI:10.32604/biocell.2021.014279
| Biocell DOI:10.32604/biocell.2021.014279 | |
| Article |
Benefit of prophylactic bronchodilator with β2 adrenergic agonist in ischemia-reperfusion-induced lung injury
1Division of Pulmonary and Critical Care, Department of Internal Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei, 114, Taiwan
2Post-Baccalaureate Program in Nursing, College of Nursing, Taipei Medical University, Taipei, 110, Taiwan
3Department of General Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei, 114, Taiwan
4Division of Pulmonary Immunology and Infectious Diseases, Chest Department, Taipei Veterans General Hospital, Taipei, 112, Taiwan
*Address correspondence to: Chi-Huei Chiang, chiang1990@gmail.com; Chih-Feng Chian, sonice3982@gmail.com
#These authors contributed equally to this work
Received: 15 September 2020; Accepted: 24 February 2021
Abstract: Primary lung graft dysfunction could significantly attribute to ischemia-reperfusion lung injury (IRLI) during transplantation surgery. β2-adrenergic agonists were one of the bronchodilators that had been well-established in the management of asthma and chronic obstructive pulmonary disease (COPD) with anti-inflammatory potency. By applying the model of isolated rat lung, we evaluated the efficacy of short-acting β2-agonist inhalation to ameliorate ischemia-reperfusion damage. The experiment protocol was 180 min of global ischemia and then reperfusion for 60 min. In the β2-agonist inhalation group, aerosolized albuterol was administrated prior ischemia procedure. Increased weight ratios of wet to dry lung and microvascular permeability were characterized in the IRLI group. In contrast, pre-inhaled β2-agonist significantly mitigated the severity of pulmonary edema. Bronchoalveolar lavage from the β2-agonist group presented decreased leukocyte counts and cytokines production, including interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and macrophage inflammatory protein 2 (MIP-2). Devastating oxidative stress was widely recognized during the ischemia-reperfusion process, while β2-agonist pretreatment revealed subsided H2O2, myeloperoxidase (MPO), and the cleavage of caspase-3. Western blotting from lung homogenates identified the blockade of NF-κB and MAPK activation in the β2-agonist inhalation group. Currently, there was no specific pharmacotherapy in IRLI management. Our results elucidated the protective effect of β2-agonist bronchodilator against ischemia-reperfusion induced oxidative stress, inflammation reaction, and pulmonary edema.
Keywords: Ischemia-reperfusion lung injury; β2-adrenergic agonist; Bronchodilator; Lung transplantation
Abbreviations
| I/R: | ischemia-reperfusion |
| IRLI: | ischemia-reperfusion lung injury |
| Kfc: | pulmonary capillary filtration coefficient |
| LWG: | lung weight gain |
| MKP-1: | mitogen-activated protein kinase phosphatase 1 |
| MPO: | myeloperoxidase |
| Ppa: | pulmonary arterial pressure |
| Ppc: | pulmonary capillary pressure |
| Ppv: | pulmonary venous pressure |
| Ra: | pulmonary arterial resistance |
| Rv: | venous resistance |
In lung transplantation, the unavoidable process of organ ischemia and subsequent reperfusion mainly contributed to acute allograft injury within the first 72 hours (Lama et al., 2017). Grossly, ischemia-reperfusion lung injury (IRLI) was characterized by pulmonary infiltration, decreased compliance, and aggressive hypoxemia (Laubach and Sharma, 2016). Histopathological examination for IRLI encompassed accumulation of protein exudation in the interstitium, infiltration of neutrophils and monocytes, as well as massive pneumocytes apoptosis (De Perrot et al., 2003). At the molecular level, there was an activation of MAPK, PI3K/Akt, and ROS signaling that collaborated to acute lung injury (Liang et al., 2019; Okada et al., 2013; Ovechkin et al., 2007). The epidemiological analysis had indicated the association between severe IRLI and decreased long-term survival of transplant operation (Christie et al., 2012). However, the standard pharmacotherapy for IRLI remains controversial, especially for prevention. From studies enrolled in ClinicalTrials.gov, only nitric oxide (NO) inhalation and repertaxin as a CXCL8 inhibitor had been investigated in patients receiving lung transplantation for reducing IRLI. As a result, the drug development of IRLI prevention should be emphasized in order to improve patient outcomes after transplantation surgery.
Short-acting β2-agonists as bronchodilators were extensively prescribed to relieve bronchospasm for decades in asthmatic attacks. Beyond its broncho-relaxing effect, literature had claimed the activation of β2 receptor to modulate inflammatory response (Padro and Sanders, 2014). One mechanism could be attributed to the abundant expression of β2 receptors on the surface of leukocytes, including neutrophils, lymphocytes, and macrophages (Uzkeser et al., 2012). The combination of β2-agonist and an anticholinergic agent was proved to inhibit neutrophils infiltration and matrix metalloproteinase-9 (MMP-9) activity in drug-induced pulmonary inflammation model (Zhang et al., 2010). In clinical trials with acute respiratory distress syndrome (ARDS), intravenous salbutamol had failed to improve mortality rate due to systemic side effects despite previous promising results in reducing lung edema (Perkins et al., 2006; Gao Smith et al., 2012). For patients undergoing lung resection, aerosolized β2-agonist and anticholinergic agent within the first 36 postoperative hours had been shown to accelerate the clearance of extravascular lung water (Licker et al., 2008). However, the potential of inhaled β2-agonist for prevention has not been explored in acute lung injury induced by ischemia-reperfusion (I/R).
The component of IRLI in primary graft dysfunction could be recapitulated by preclinical models, including unilateral hilar occlusion, isolated, perfused rodent lungs, and orthotopic lung transplantation (Lama et al., 2017). Key indexes to assess these models included cardiopulmonary hemodynamics, the gravimetric (wet/dry) ratio for lung edema, and the protein analysis of bronchoalveolar lavage (Matute-Bello et al., 2011; Sayah et al., 2015). The potency of β2-agonist inhalation in anti-inflammation prompted us to investigate its prophylaxis use in IRLI. Therefore, the present study aimed to evaluate the benefit of the bronchodilator albuterol in an isolated ex vivo model through mitigating leukocyte infiltration, oxidative stress, and microvascular permeability.
The study protocol was approved by the Institutional Review Board of Taipei Veterans General Hospital Subcommittee for the Care of Animal Subjects. Animal care and handling practices were in accordance with the National Institutes of Health guidelines for ethical animal research. The model of the isolated perfused lung in situ I/R was described previously (Lu and Chiang, 2008). Briefly, male Sprague–Dawley rats (body weight: 250–350 g) were anesthetized with an intraperitoneal injection of sodium pentobarbital. Subsequently, a tracheotomy was performed, and mechanical ventilation was applied (Rodent Ventilator Model 683; Harvard Apparatus, South Natick, MA, USA) at a tidal volume of 5 mL/kg and a positive end-expiratory pressure of 2 cmH2O.
After sternotomy, heparin (1 unit/g) was injected into the right ventricle through which the pulmonary artery was catheterized. The left atrium was also catheterized. The pulmonary venous outflow was diverted into a reservoir. To prevent backflow into the ventricles, an additional ligation was performed above the atrioventricular junction. The lungs were then perfused with 10 mL of blood mixed with 20 mL of 0.9% normal saline (Minipulse 2; Gilson Medical Electronics, Middleton, WI, USA) at a constant flow rate of 30 mL/min/g body weight. The pulmonary arterial pressure (Ppa), pulmonary venous pressure (Ppv), and peak airway pressure were monitored using pressure transducers (P23 ID; Statham, Oxford, CA, USA) and recorded on a polygraph (Gould Instruments, Cleveland, OH, USA). The weight of each rat was determined to reflect the lung weight in situ, and the lung weight gain (LWG) was recorded continuously.
Determination of pulmonary capillary pressure (Ppc)
The Ppc was estimated using the double occlusion method. The arterial inflow and venous outflow lines were occluded simultaneously, and the equilibrium Ppa and Ppv were measured. The equilibration pressure correlated well with the isogravimetric measurements of Ppc and reflected the prevailing capillary pressure when the lung was not isogravimetric.
The pulmonary arterial resistance (Ra) and venous resistance (Rv) were calculated using the following equations: Ra = (Ppa–Ppc)/Q and Rv = (Ppc–Ppv)/Q, where Q represents the perfusate flow.
Measurement of microvascular permeability
The pulmonary capillary filtration coefficient (Kfc) was used as an index of the microvascular permeability to water. Kfc was measured using a previously described method (Chiang et al., 2008). Briefly, the Ppv was elevated rapidly to 6–8 cm H2O for 15 min after an isogravimetric period. The consequent increase in lung weight was recorded. A characteristic period of rapid weight gain (i.e., vascular filling) was followed by a period of slower weight gain. The rate of weight change (DWt/Dt) during the 6- to 14-min interval was analyzed using linear regression of the log10-transformed rates of weight change per minute. The initial weight gain rate was calculated using an extrapolation of DWt/Dt to time 0. Next, Kfc was calculated by dividing the DWt/Dt at time 0 by the change in Ppc after the venous outflow pressure was increased. After normalization using the baseline wet lung weight, the Kfc was expressed in units of mL/min/cm H2O per 100 g of lung tissue.
The experiment was initiated after hemodynamic stability had been attained for 15 min in the extracorporeal isolated lung circulation system. Rats were divided into three treatment groups—control, I/R, and I/R+β2 agonist—and all groups were subjected to isolated lung preparations ventilated with tidal volume settings at 5 mL/kg. I/R injuries were induced according to the following protocol: ventilation and perfusion of the isolated lung were discontinued for 180 min (ischemia) and then reinstituted (reperfusion) for 60 min at room temperature. Prior to ischemia, rats in the I/R+β2 agonist group were subjected to an inhaled β2-agonist treatment (2-puff dose using Ventolin Meter Dose Inhaler, 100 μg albuterol/inhalation; GlaxoSmithKline, Middlesex, UK) delivered by an in-line spacer adapted to the inspiratory limb of the ventilator circuit. The spacer was kept in line for 60 s after each actuation (2 actuations).
White blood cell (WBC) count in the bronchoalveolar lavage fluid (BALF)
All experiments were terminated after closed extracorporeal perfusion. The lungs were removed, and the wet weights were measured. The lungs were then lavaged twice by instilling saline (2.5 mL/lavage) into the left upper lobe. Lavage samples were centrifuged at 1500×g and at room temperature for 10 min to determine the white blood cell (WBC) counts.
The concentration of myeloperoxidase (MPO), an index of neutrophil sequestration, was measured in the lungs, as previously described (Chiang et al., 2008).
The perfusate was centrifuged at 1000×g within 30 min, and the supernatant was collected into 50 mL of H2O2 Reaction Mix containing 46 mL of Assay buffer and 2 mL of OxiRedTM Probe solution. Two milliliters of HRP solution (BioVision, Linda Vista Avenue Mountain View, CA, USA) were added, and the mixture was incubated for 10 min. The absorbance was then read at 570 nm (SpectraMax M5, Molecular Devices, Sunnyvale, CA, USA). The concentration was then calculated based on H2O2 standard curves.
The concentrations of interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and macrophage inflammatory protein 2 (MIP-2) in BALF were measured using commercial enzyme-linked immunosorbent assay kits (R&D Systems, Oxon, UK). For each assay, absorbance in each well was read at 450 nm (SpectraMax M5; Molecular Devices, Silicon Valley, CA, USA).
Lung tissues were homogenized in a lysis buffer containing protease inhibitor cocktail (Roche, Indianapolis, IN, USA) and phosphatase inhibitor cocktail (Roche). Total protein extracts were separated on 10% sodium dodecyl sulfate-polyacrylamide gels and electro-transferred onto PVDF membranes (Millipore, Billerica, MA, USA). The membranes were then blocked with 5% non-fat dry milk in Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBST) for 1 h. The following primary antibodies were used: phospho-p44/42 MAPK (p-ERK1/2), phospho-SAPK/JNK (p-JNK), phospho-p38 MAPK (p-P38), anti-p44/42 MAPK (ERK1/2), anti-SAPK/JNK (JNK), and anti-p38 MAP Kinase (P38) (1:1000 dilution; Cell Signaling Technology, Beverly, MA, USA); Caspase-3, P-AKT, AKT, PAI-1, and AP-1 (1:2000; Cell Signaling Technology); and GADPH (1:10000; Lab Frontier). Subsequently, an appropriate secondary antibody was used (horseradish peroxidase anti-rabbit IgG, 1:10000; Jackson Immuno Research Laboratories, West Grove, PA, USA). Labeled protein bands were visualized using enhanced chemiluminescence (Visual Protein Biotechnology Crop, Taiwan) and quantified using Kodak 1D Image Analysis Software (Eastman Kodak Company, Rochester, NY, USA).
NF-κB analysis of nuclear protein
Lung tissues were homogenized in 5 mL of solution A (0.6% Nonidet P-40, 150 mmol/L NaCl, 10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.9, 1 mmol/L ethylenediaminetetraacetic acid [EDTA], and 0.5 mmol/L phenylmethylsulfonyl fluoride [PMSF]) using a Dounce tissue homogenizer. The homogenates were centrifuged for 30 s at 2000 rpm, and the supernatants were then collected and centrifuged again for 5 min at 5000 rpm. The pelleted nuclei were resuspended at 4°C in 300 µL of solution B (25% glycerol, 20 mmol/L HEPES, pH 7.9, 420 mmol/L NaCl, 1.2 mmol/L MgCl2, 0.2 mmol/L EDTA, 0.5 mmol/L dithiothreitol, 0.5 mmol/L PMSF, 2 mmol/L benzamidine, 5 mg/mL pepstatin A, 5 mg/mL leupeptin, and 5 mg/mL aprotinin) and incubated on ice for 20 min. After the samples were centrifuged at 15000 rpm for 1 min, the total protein concentration in each extract was determined using a BCA protein assay (Pierce, Rockford, IL, USA). The membrane was blocked for 1h. Antibodies specific for NF-κB (1:1000; Cell Signaling Technology) and proliferating cell nuclear antigen (PCNA) (1:1000; Cell Signaling Technology) were diluted in TBST buffer and incubated overnight at 4°C. The appropriate secondary antibody was used (1:10000 horseradish peroxidase anti-rabbit) at room temperature for 1 h. Visualization was achieved using enhanced chemiluminescence (Visual Protein Biotechnology Corp, Taipei). Anti-PCNA antibody was used as a loading control to correct for the pixel values corresponding to NF-κB.
Lung histopathology and lung injury score
After the termination of each experiment, lung tissue in the right lower lobes was dissected and fixed immediately in 10% neutral buffered formalin. After fixation, the lung tissues were dehydrated through a graded series of alcohol, cleared in xylene, and embedded in paraffin. All sections were cut to 5 mm and stained with hematoxylin/eosin. The severity of perivascular, peribronchial, septal, and alveolar edema as well as perivascular, interstitial, and alveolar cell infiltration was examined by a scoring system. We used lung pathology score as previously developed (Chiang et al., 2011). In brief, the scoring method measure the severity of acute lung injury: perivascular edema = 1; peribronchial edema = 2; interstitial edema = 2; alveolar edema = 3; perivascular cell infiltration = 2; interstitial cell infiltration = 3; alveolar cell infiltration = 4. A total of 20 scope views were examined for each lung tissue specimen. The sum of all the pathological scores was the score for each scope, and then we calculated the mean score of 20 scopes as the injury score for this lung tissue. Blind reviews were carried out by 2 pathologists, and the mean of these 2 scores was taken as the final score.
Systat10.0 (Systat Software Inc, San Jose, CA, USA) was used for the statistical analysis. Comparisons among all groups were conducted using an analysis of variance, followed by Dunnett’s post hoc test. Comparisons between the baseline control and post-I/R values within each group were conducted using a paired Student’s t-test. Values are expressed as means ± standard deviations (SD). A P-value of <0.05 was considered statistically significant.
Effects of β2-agonist inhalation on pulmonary hemodynamic parameters
Rats were randomly assigned as (a) control group ventilated with tidal volume (VT) of 5 mL/kg, (b) IRLI group underwent 180 min of global ischemia and then reperfusion for 60 min, and (c) β2-agonist inhalation group treated prior to I/R period. β2-agonist was prepared with aerosolized albuterol of 200 μg and administrated through an in-line spacer connected to the inspiratory limb of the ventilator circuit. After the I/R period, pulmonary hemodynamic variables were not statistically different by β2-agonist inhalation (Tab. 1).
Effect of β2-agonist inhalation against pulmonary edema and inflammation
Lung wet gain (LWG) served as an indicator of pulmonary edema, as well as pulmonary capillary filtration coefficient. The IRLI group showed a significant increase in the ratio of the lung to body weight and microvascular permeability (Tab. 2). Compared with the IRLI group, β2-agonist inhalation significantly suppressed the severity of pulmonary edema from 3.33 ± 1.41 to 1.11 ± 0.72, respectively, in LWG (P < 0.05). From the analysis of BALF, leukocyte count, cytokines production, and H2O2 level reflected pulmonary inflammatory status. I/R exerted leukocytes infiltration, whereas the β2-agonist inhalation group revealed fewer leukocyte counts (Tab. 2).
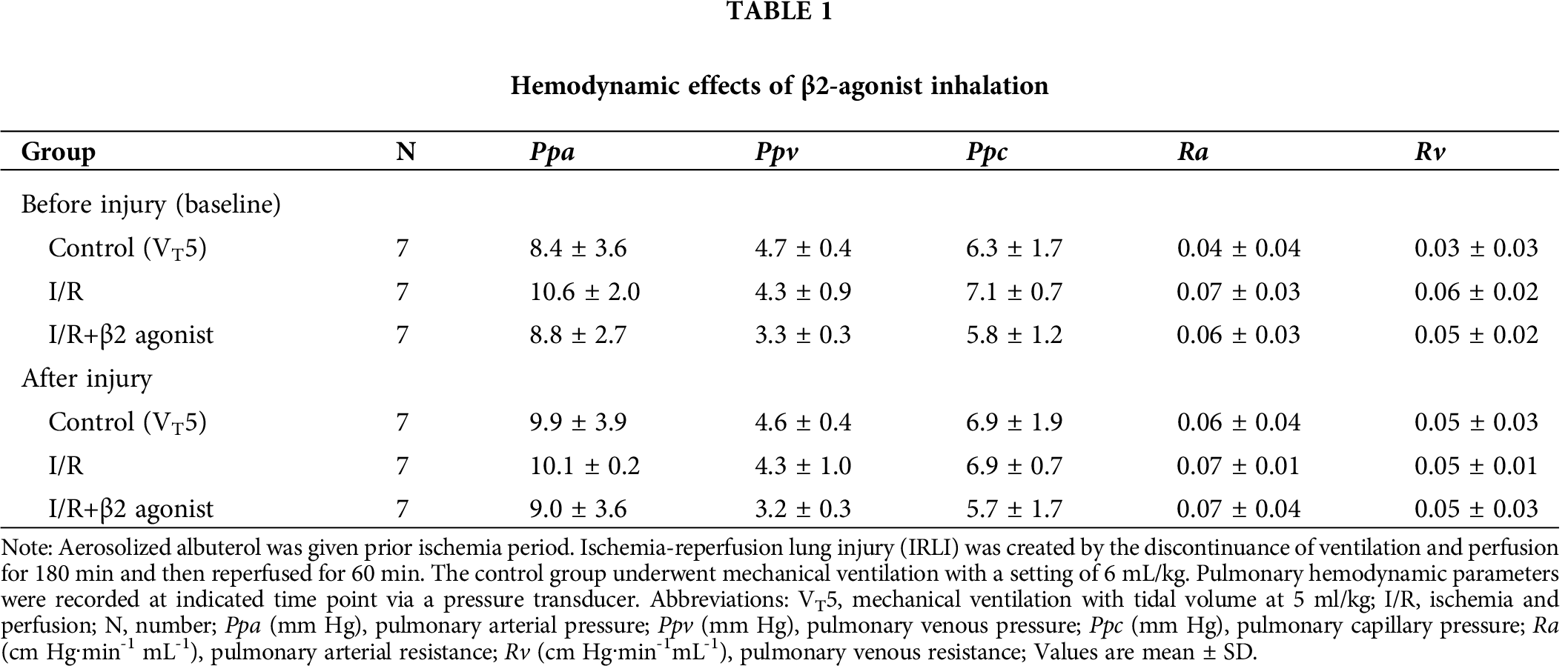
Moreover, the H2O2, MIP-2, IL-1β, and TNF-α concentrations in lavage fluid were higher in the I/R group than in the control group (Fig. 1). The inhalation of β2 agonist largely attenuated the oxidative stress and cytokine responses (Fig. 1). Consistent with BALF results, histopathological examination of the β2-agonist inhalation group identified significantly decreased interstitial and airspace leukocyte infiltration (Fig. 2). In addition, pretreated with β2-agonist alleviated alveolar septal thickening, intra-alveolar leukocytic infiltrates, and lung injury score observed in the IRLI group (Fig. 2E). Myeloperoxidase (MPO) had a positive correlation with neutrophil infiltration. Compared with the IRLI group, MPO concentration was lower in the β2-agonist pretreated group (Fig. 2).
Next, we examined protein expression in lung homogenates that also showed decreased IL-1β and TNF-α levels (Figs. 3A and 3B). Plasminogen activator inhibitor-1 (PAI-1) could be another index of lung oxidative stress. There was a non-significant difference between I/R and β2-agonist inhalation group (Fig. 3C). These findings indicate that the inhalation of β2 agonist largely attenuated I/R-induced oxidative stress, cytokine responses, and pulmonary edema.
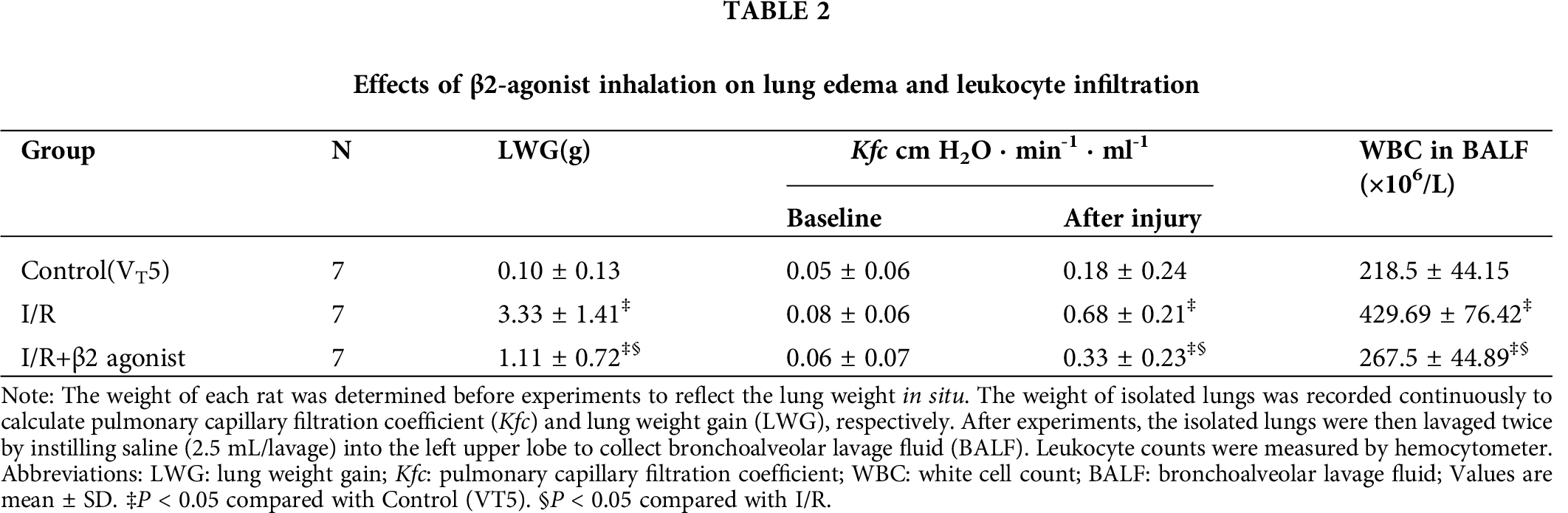
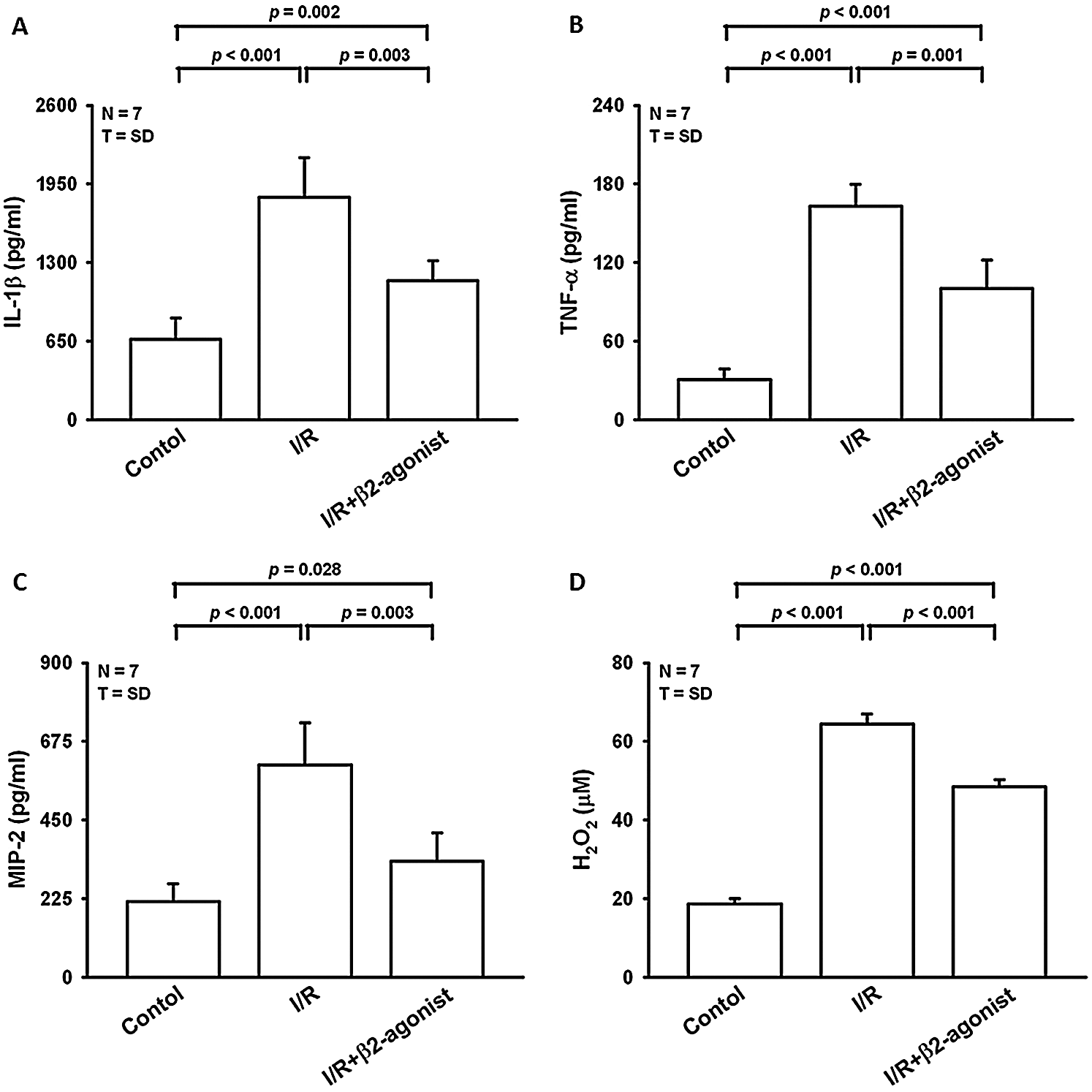
Figure 1: Effect of β2-agonist inhalation on the composition of cytokines and H2O2 in BALF. Levels of (A) interleukin-1β (IL-1β), (B) tumor necrosis factor-α (TNF-α), (C) macrophage inflammatory protein 2 (MIP-2) from BALF were determined by ELISA. (D) H2O2 level in BALF was measured by a colorimetric method. T represents standard deviation. N, number of animals used.
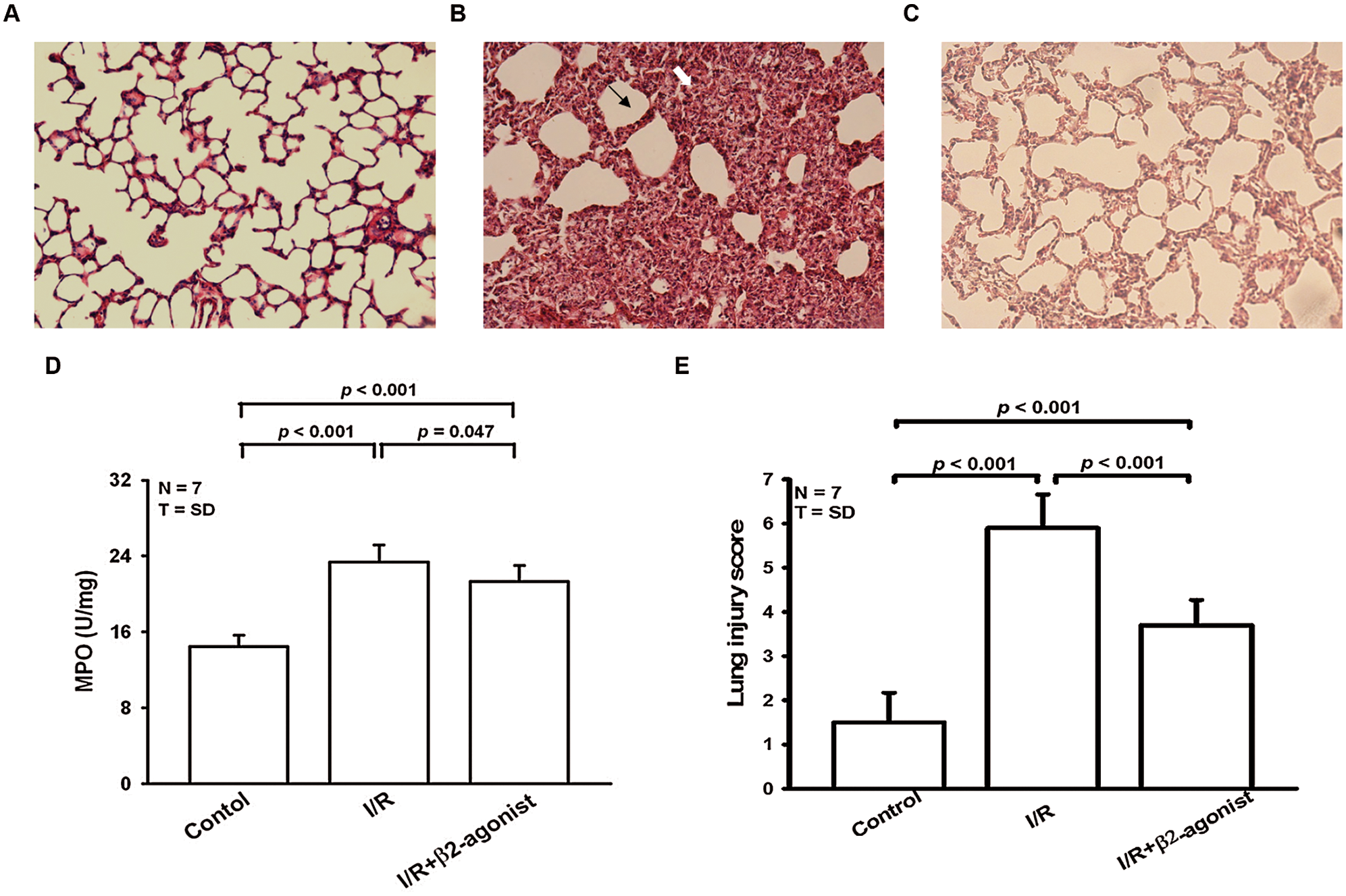
Figure 2: β2-agonist inhalation alleviated histopathological changes, myeloperoxidase (MPO) elevation, and lung injury score. Histological analysis (HE-stain, 100×) of lung tissues from (A) control group, (B) I/R group, and (C) pretreated β2 agonist group. (D) Levels of myeloperoxidase (MPO) from lung tissues were determined by ELISA. (E) Pathologic lung injury score. Black arrow: septal thickening. White arrow: intra-alveolar leukocyte infiltrates. T represents standard deviation. N, number of animals used.
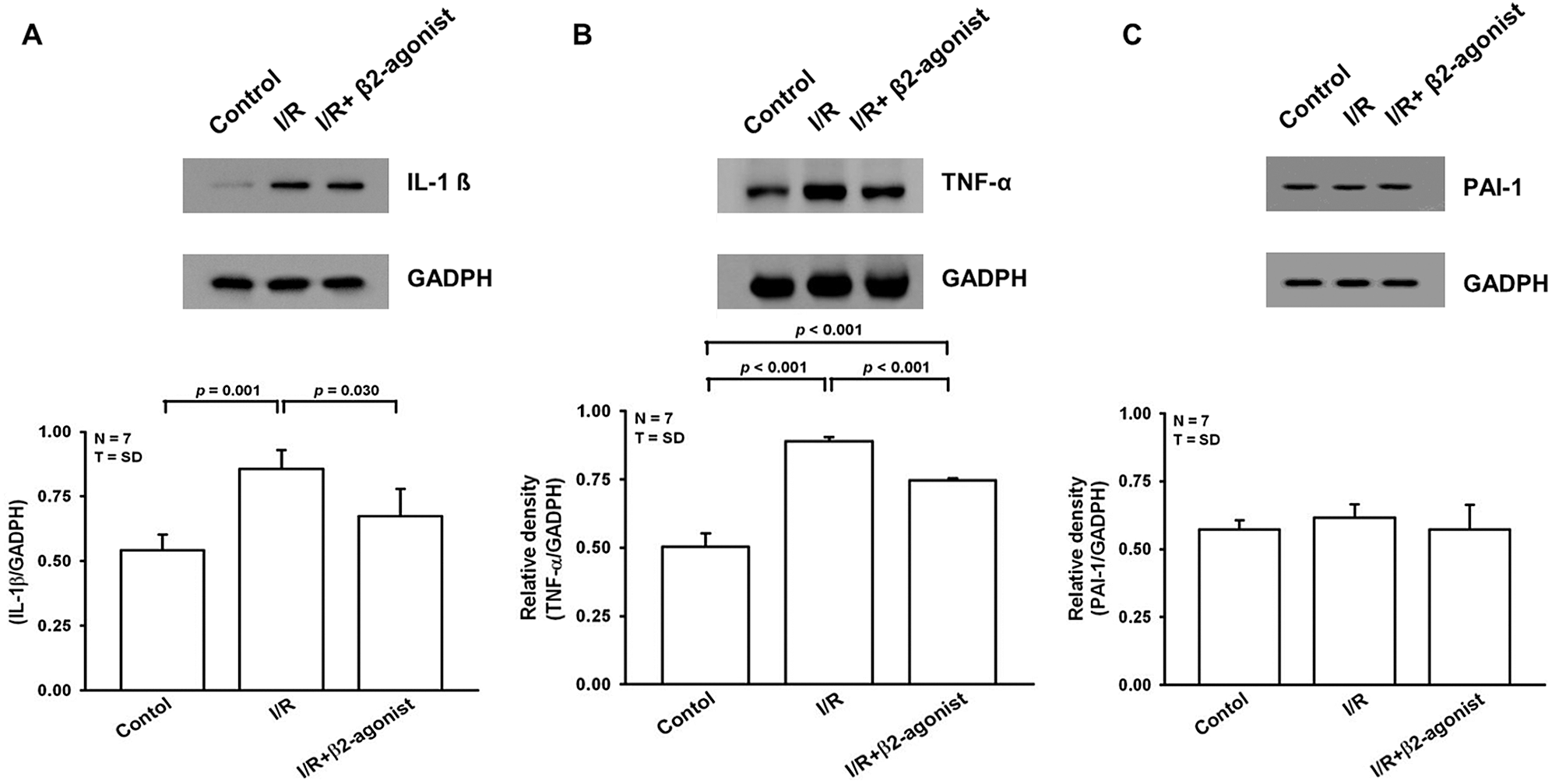
Figure 3: Effect of β2-agonist inhalation on IL-1β, TNF-α, and plasminogen activator inhibitor-1 (PAI-1). Western blot analysis of (A) IL-1β, (B) TNF-α, and (C) PAI-1 in lung tissue homogenates. T represents standard deviation. N, number of animals used.
Decreased apoptosis and NF-κB activation in β2-agonist inhalation group
Oxidative stress during I/R has been observed to trigger cell apoptosis. Western blotting showed elevated cleavage caspase-3 expression in the IRLI group that was suppressed in the β2-agonist pretreated group (Fig. 4A). The pathogenic role of NF-κB was widely documented in acute lung injury. An analysis of nuclear proteins from lung tissue revealed upregulated NF-κB expression was observed in the I/R group and lower expression in the β2-agonist treatment group (Fig. 4B). Furthermore, β2-agonist pretreatment could reverse I/R-induced NF-κB nuclear translocation. As a result, β2-agonist bronchodilator had the potency to inhibit apoptosis signaling and NF-κB activation in the IRLI model.
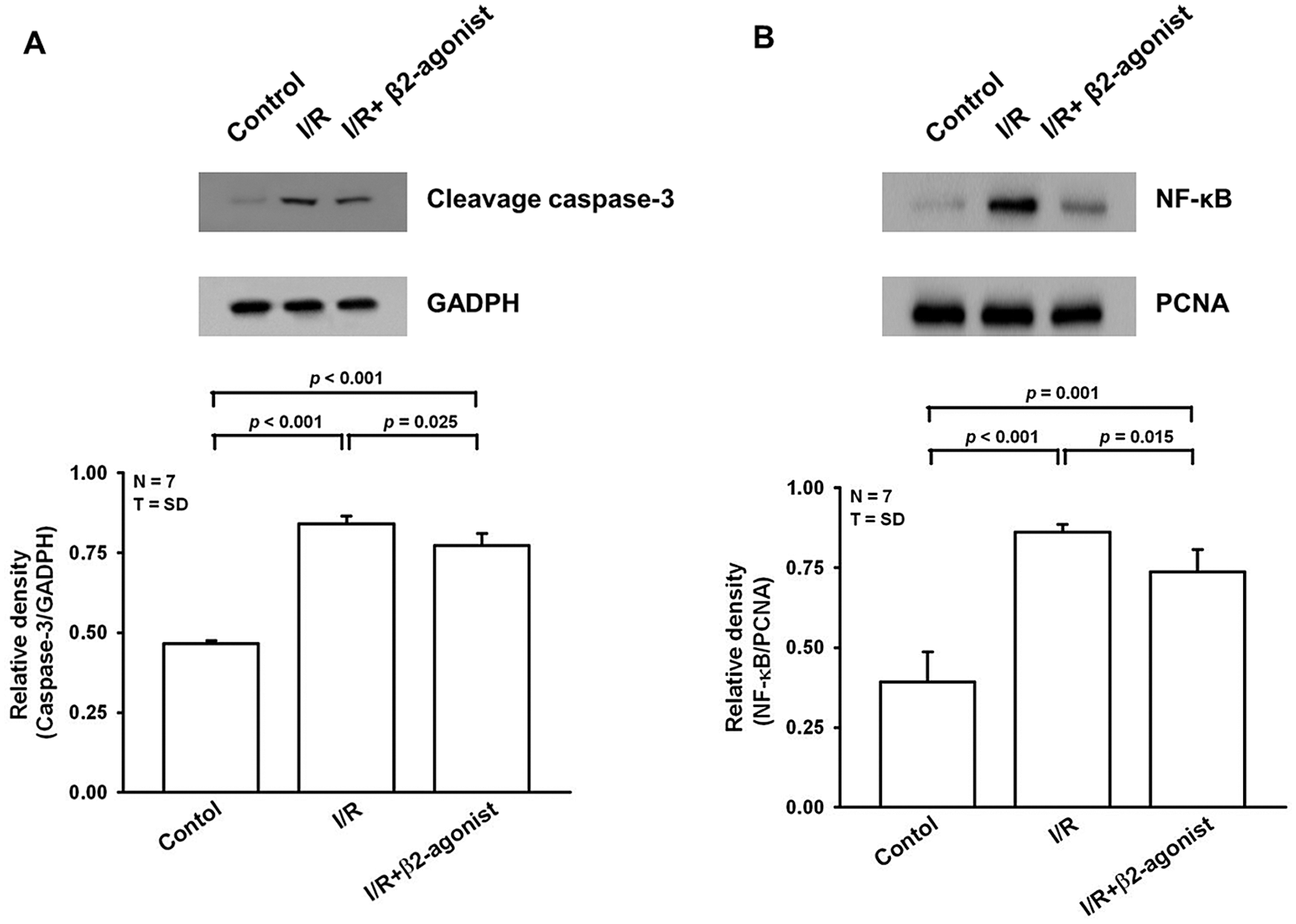
Figure 4: β2-agonist inhalation prevented apoptosis and down-regulated NF-κB activation. Western blot analysis of (A) cleavage caspase-3nuclear and (B) NF-κB and in lung tissues homogenates. GAPDH and proliferating cell nuclear antigen (PCNA) were used as the internal control, respectively. T represents standard deviation. N, number of animals used.
Blockade of MAPK phosphorylation by β2-agonist pretreatment
At the cellular level, marked activation of MAPK signaling pathways reflected the facilitation of cell migration that has been correlated with primary lung graft dysfunction. There was elevated phosphorylation of p38, ERK, and JNK in lung tissue homogenates from the IRLI group (Figs. 5A–5C). β2-agonist pretreatment generally mitigated I/R-induced MAPK activation. PI3K/Akt pathway was crucial to cell survival that could function as a protective mechanism during I/R. Increased Akt phosphorylation was observed in both I/R and β2-agonist inhalation groups without a statistical difference (Fig. 5D). Conclusively, the therapeutic mechanism of β2-agonist was postulated through inhibiting MAPK phosphorylation with protective Akt expression.
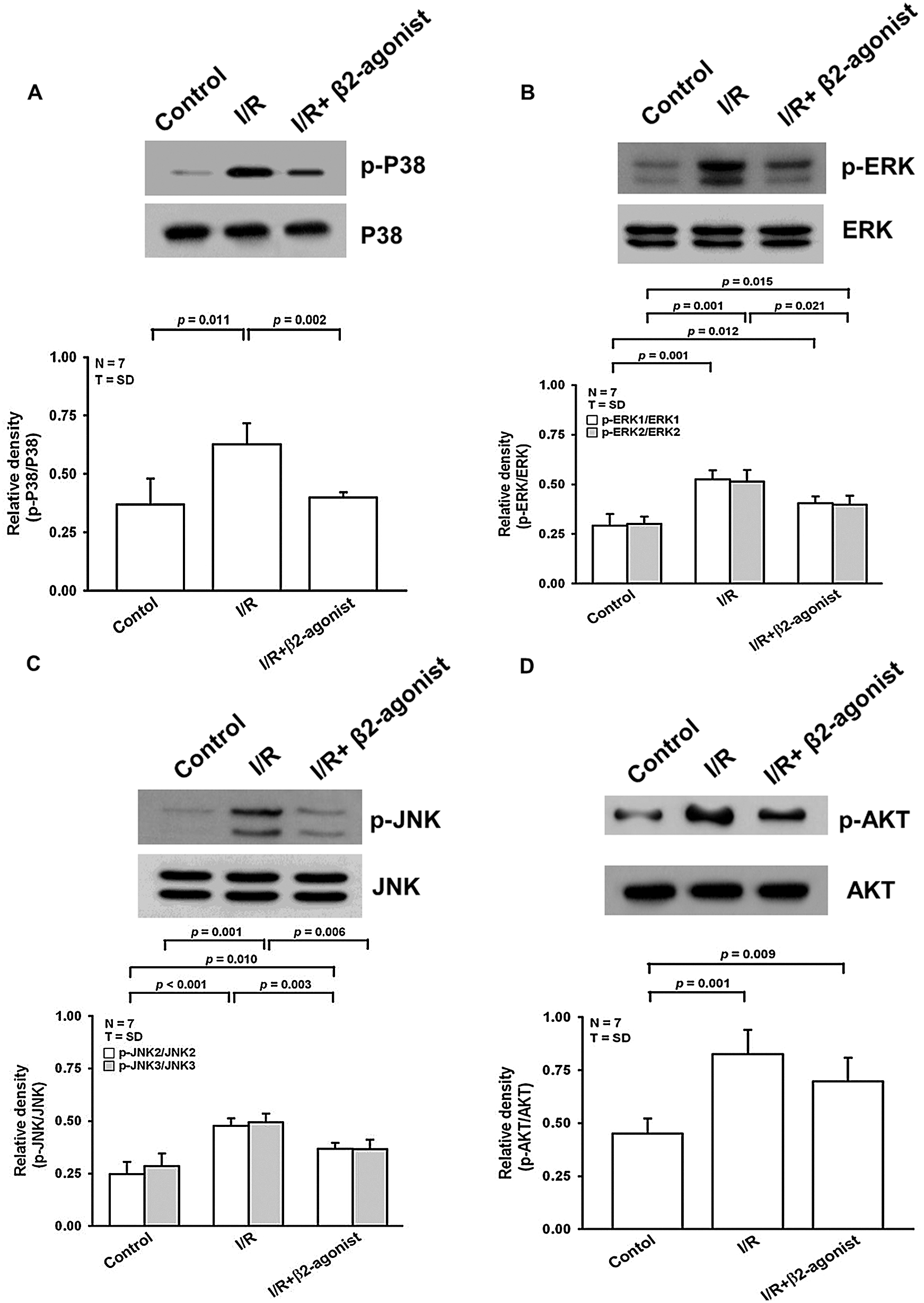
Figure 5: Effect of β2-agonist inhalation on MAPKs and AKT activation. Western blot analysis of (A) p-ERK, (B) p-JNK, (C) p-p38, and (D) p-AKT in lung tissue homogenates. T represents standard deviation. N, number of animals used.
In this study, we demonstrated that lung injury induced by a 3-hour period of ischemia and a 1-hour period of reperfusion led to increased pulmonary vascular permeability, inflammatory cell infiltration, pulmonary edema, cytokine responses, MAPK activation, NF-κB translocation, and apoptotic enzymes (caspase-3 and p-AKT) expression. Moreover, inhalation of β2 receptor agonist prior to ischemia inhibited MAPK activation, suppressed NF-κB activation, reduced inflammatory cytokines release and hydrogen peroxide production, and attenuated apoptotic responses (Fig. 6).
Thus far, randomized clinical trials have failed to demonstrate the efficacy of β2 agonists in the attenuation of acute respiratory distress syndrome (ARDS). In the Albuterol Treatment for Acute Lung Injury (Mitchell et al., 2011) study, which examined whether a 10-day regimen of high-dose aerosolized albuterol would facilitate alveolar fluid clearance in 282 patients with ARDS (Heart, 2011), the ALTA study not only failed to demonstrate positive results but also suggested several side effects, including tachycardia, cardiac arrhythmias, and hypokalemia. The injured alveolar epithelium of subjects in the ALTA study might not be capable of responding to β2 agonist stimulation. Additionally, the dosage could be distributed unevenly between healthy and injured regions of the lung, leading to cardiovascular effects associated with higher β2 concentrations. In contrast to past studies which administered β2 agonist after ischemia injury, we preconditioned the lungs with short-acting β2 agonist inhalation. Furthermore, our use of short-acting inhalation drug could reduce the incidence of systemic effects observed in other studies of intravenously administered β2 agonist, such as the BALTI-2 (Beta Agonist Lung Injury Trial-2) and BOLD (Beta-agonists for Oxygenation in Lung Donors) studies (Gao Smith et al., 2012; Ware et al., 2014). In our study, the inhalation of β2 agonist prior to ischemia induction reduced the IR-associated gains in lung wet weight and Kfc, thus preventing edema associated with 3-hour lung I/R.
Several factors might explain why β2 adrenergic aerosol therapy improved the outcomes of model animals in our study but did not yield evident benefits in clinical trials. First, our study administered aerosolized albuterol to non-edematous alveoli, in contrast to clinical trials in which β2-agonists were given after injury. The response of healthy epithelium to β2-agonist was quite different from those subjects with existing ALI/ARDS that were characterized by apoptotic and necrotic debris. A comparison of these results suggests that a relatively intact barrier of alveolar epithelium is required for pulmonary fluid clearance.
Second, the observed differences may be attributable to the ex vivo animal model, which might not harbor the same risk of cardiovascular effects as human subjects. However, the BOLD study reported apparent improvements in the long-term prognosis of heart transplant recipients treated with albuterol when compared to those without albuterol treatment. This cardiovascular benefit might be associated with β2 agonist-mediated changes in endogenous catecholamine production or inflammatory cascade modulation (Maris et al., 2005).
Despite the lack of support from clinical trials, significant evidence in the literature supports the findings of the present study. Several studies have suggested that β2 agonists reduced endothelial damage and enhanced repair in lung injury models (Sakamoto et al., 2012; Cepkova and Matthay, 2006). Moreover, in vitro studies demonstrating the ability of β2 agonists to stimulate the closure of mechanically induced wounds in epithelial monolayers by increasing cAMP and activating protein kinase A (Gropp et al., 2011) provide evidence supporting the role of these agents in epithelial repair (Perkins et al., 2008).
Elevated intracellular cAMP levels, which protected lungs against injury, decreased under hypoxic conditions (Matthay, 2014; Matthay and Abraham, 2006; Vivona et al., 2001). The clinical importance of cAMP role was underscored by Chen and colleagues, who demonstrated that nebulized salmeterol, a long-acting β2 agonist, maintained cAMP levels in the lung and alleviated pulmonary I/R injury (Chen et al., 2006; Hoffmann et al., 2001). These findings suggest that the protective effects observed in this study might be attributable to albuterol-mediated activation of β-adrenoceptor, a transmembrane G-protein-coupled receptor that activates adenylate cyclase (AC), leading to increases in intracellular cAMP.
Our results also suggest that the protection provided by albuterol against IRLI was mediated partially through the anti-inflammatory effects of this agent. Specifically, we found that inhalation of a β2 agonist reduced oxidative stress, suppressed IR-related proinflammatory cytokine production, attenuated NF-κB activation, and decreased p38, ERK, and JNK MAPK phosphorylation. These findings are consistent with previous studies demonstrating that albuterol increases mitogen-activated protein kinase phosphatase 1 expression (MKP-1) and suppresses p38 MAPK phosphorylation, while adrenaline induces the expression of genes encoding anti-oxidative factors, such as nuclear factor E2 p45-related factor-2 (Nrf2), and thus guards against oxidative stress (Keränen et al., 2016; Takahata et al., 2009). Thus, our findings extend the previous body of knowledge by providing ex vivo evidence that MAPK at least partly mediates the anti-inflammatory effects of albuterol.
In conclusion, we found that the inhalation of adrenergic β2 agonist prior to ischemia reduced the injury associated with reperfusion by reducing MAPK activation and lung tissue apoptosis. Accordingly, preoperative β2 agonist therapy might help to reduce IRLI associated with lung transplantation.
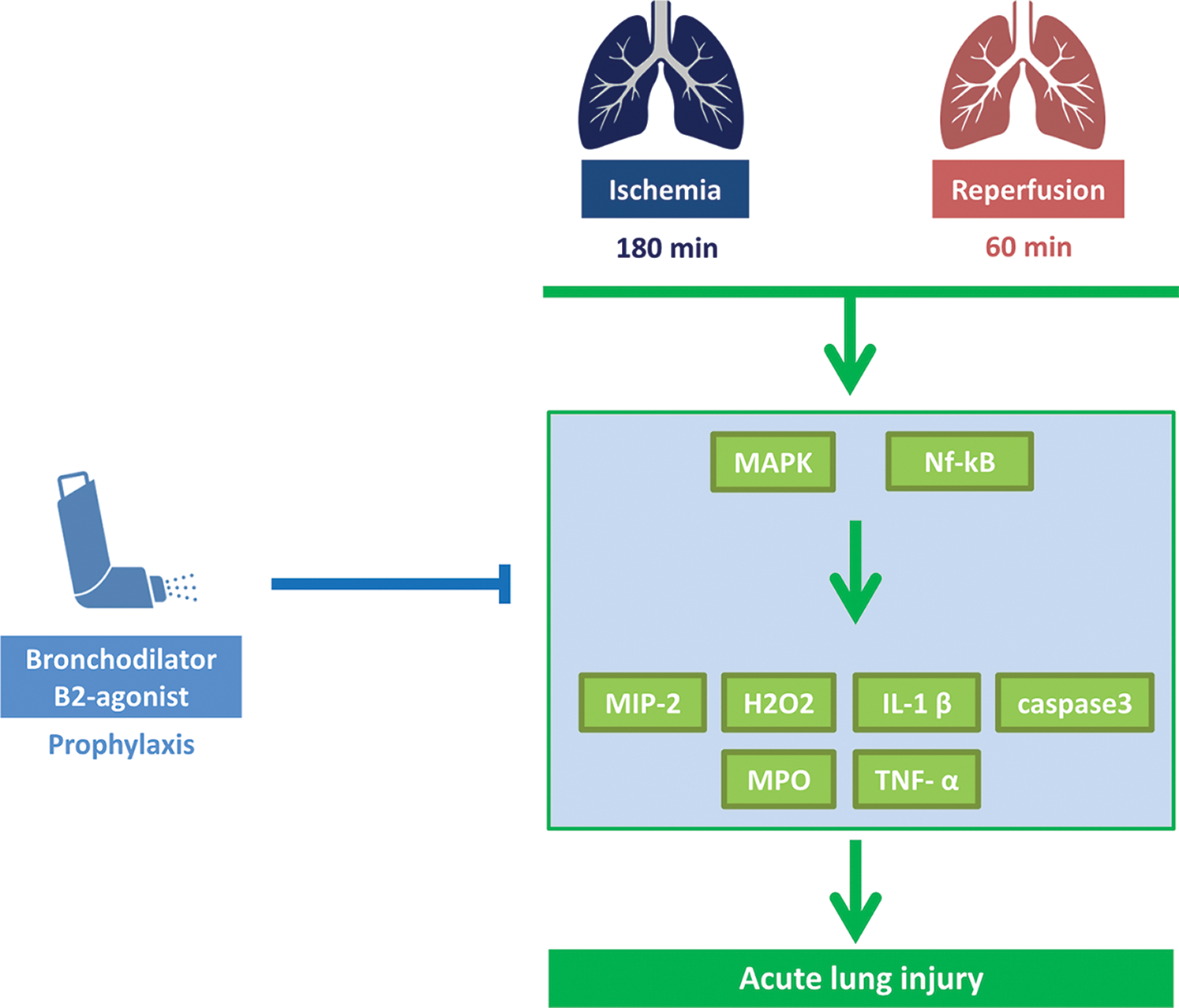
Figure 6: Scheme of IRLI protected by β2-agonist inhalation. Prophylactic inhalation with β2-agonist could ameliorate acute lung injury induced by ischemia-reperfusion through inhibited MAPK activation, suppressed NF-κB nuclear translocation, reduced inflammatory cytokines release, and hydrogen peroxide production, and attenuated apoptotic responses.
Perspective Statement: The ischemia-reperfusion process greatly compromised the outcomes of lung transplantation. Short-acting β2-agonist has been widely used in the emergent management of patients with asthma. Moreover, bronchodilators possessed excellent biosafety and cost-effectiveness. The present study was thus aimed to evaluate the potency of preventive β2-agonist in the animal models of ischemia-reperfusion lung injury.
Authors’ Contributions: CFC conceived and designed the experiments. CHC performed the experiments CLT, YHL and CYC wrote the manuscript. CFC and CHC contributed to the project’s design and implementation, collecting funds, and revising the manuscript thoroughly. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All the data supporting these findings is contained within this manuscript.
Funding Statement: This work was supported by the Tri-Service General Hospital, National Defesnse Medical Center in Taiwan (TSGH-C108-109).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present review.
Cepkova M, Matthay MA (2006). Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. Journal of Intensive Care Medicine 21: 119–143. DOI 10.1177/0885066606287045. [Google Scholar] [CrossRef]
Chen F, Nakamura T, Fujinaga T, Zhang J, Hamakawa H, Omasa M, Sakai H, Hanaoka N, Bando T, Wada H, Fukuse T (2006). Protective effect of a nebulized β2-adrenoreceptor agonist in warm ischemic-reperfused rat lungs. Annals of Thoracic Surgery 82: 465–471. DOI 10.1016/j.athoracsur.2006.01.010. [Google Scholar] [CrossRef]
Chiang CH, Chuang CH, Liu SL (2011). Apocynin attenuates ischemia-reperfusion lung injury in an isolated and perfused rat lung model. Translational Research 158: 17–29. DOI 10.1016/j.trsl.2011.02.002. [Google Scholar] [CrossRef]
Chiang CH, Pai HI, Liu SL (2008). Ventilator-induced lung injury (VILI) promotes ischemia/reperfusion lung injury (I/R) and NF-kappaB antibody attenuates both injuries. Resuscitation 79: 147–154. DOI 10.1016/j.resuscitation.2008.02.028. [Google Scholar] [CrossRef]
Christie JD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Dobbels F, Kirk R, Rahmel AO, Stehlik J, Hertz MI (2012). The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart-lung transplant report—2012. Journal of Heart and Lung Transplantation 31: 1073–1086. DOI 10.1016/j.healun.2012.08.004. [Google Scholar] [CrossRef]
De Perrot M, Liu M, Waddell TK, Keshavjee S (2003). Ischemia-reperfusion–induced lung injury. American Journal of Respiratory and Critical Care Medicine 167: 490–511. DOI 10.1164/rccm.200207-670SO. [Google Scholar] [CrossRef]
Gropp K, Weber N, Reuter M, Micklisch S, Kopka I, Hallström T, Skerka C (2011). beta(2)-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood 118: 2774–2783. DOI 10.1182/blood-2011-02-339564. [Google Scholar] [CrossRef]
Hoffmann SC, Bleiweis MS, Jones DR, Paik HC, Ciriaco P, Egan TM (2001). Maintenance of cAMP in non-heart-beating donor lungs reduces ischemia-reperfusion injury. American Journal of Respiratory and Critical Care Medicine 163: 1642–1647. DOI 10.1164/ajrccm.163.7.9911060. [Google Scholar] [CrossRef]
Keränen T, Hömmö T, Hämäläinen M, Moilanen E, Korhonen R (2016). Anti-inflammatory effects of β2-receptor agonists salbutamol and terbutaline are mediated by MKP-1. PLoS One 11: e0148144. DOI 10.1371/journal.pone.0148144. [Google Scholar] [CrossRef]
Lama VN, Belperio JA, Christie JD, El-Chemaly S, Fishbein MC, Gelman AE, Hancock WW, Keshavjee S, Kreisel D, Laubach VE, Looney MR, McDyer JF, Mohanakumar T, Shilling RA, Panoskaltsis-Mortari A, Wilkes DS, Eu JP, Nicolls MR (2017). Models of lung transplant research: A consensus statement from the National Heart, Lung, and Blood Institute workshop. JCI Insight 2: e93121. DOI 10.1172/jci.insight.93121. [Google Scholar] [CrossRef]
Laubach VE, Sharma AK (2016). Mechanisms of lung ischemia-reperfusion injury. Current Opinion in Organ Transplantation 21: 246–252. DOI 10.1097/MOT.0000000000000304. [Google Scholar] [CrossRef]
Liang S, Wang Y, Liu Y (2019). Dexmedetomidine alleviates lung ischemia-reperfusion injury in rats by activating PI3K/Akt pathway. European Review for Medical and Pharmacological Sciences 23: 370–377. [Google Scholar]
Licker M, Tschopp JM, Robert J, Frey JG, Diaper J, Ellenberger C (2008). Aerosolized salbutamol accelerates the resolution of pulmonary edema after lung resection. Chest 133: 845–852. DOI 10.1378/chest.07-1710. [Google Scholar] [CrossRef]
Lu HL, Chiang CH (2008). Combined therapy of pentastarch, dexamethasone, and dibutyryl-cAMP or beta 2-agonist attenuates ischaemia/reperfusion injury of rat lung. Injury 39: 1062–1070. DOI 10.1016/j.injury.2007.10.022. [Google Scholar] [CrossRef]
Maris NA, De Vos AF, Dessing MC, Spek CA, Lutter R, Jansen HM, van der Zee JS, Bresser P, van der Poll T (2005). Antiinflammatory effects of salmeterol after inhalation of lipopolysaccharide by healthy volunteers. American Journal of Respiratory and Critical Care Medicine 172: 878–884. DOI 10.1164/rccm.200503-451OC. [Google Scholar] [CrossRef]
Matthay MA (2014). Resolution of pulmonary edema. Thirty years of progress. American Journal of Respiratory and Critical Care Medicine 189: 1301–1308. DOI 10.1164/rccm.201403-0535OE. [Google Scholar] [CrossRef]
Matthay MA, Abraham E (2006). β-Adrenergic agonist therapy as a potential treatment for acute lung injury. American Journal of Respiratory and Critical Care Medicine 173: 254–255. DOI 10.1164/rccm.rccm2511003. [Google Scholar] [CrossRef]
Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS (2011). An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. American Journal of Respiratory Cell and Molecular Biology 44: 725–738. DOI 10.1165/rcmb.2009-0210ST. [Google Scholar] [CrossRef]
Mitchell J, Lai LP, Peralta F, Xu Y, Sugamori K (2011). β2-Adrenergic receptors inhibit the expression of collagen type II in growth plate chondrocytes by stimulating the AP-1 factor Jun-B. American Journal of Physiology-Endocrinology and Metabolism 300: E633–639. DOI 10.1152/ajpendo.00515.2010. [Google Scholar] [CrossRef]
Okada M, Yamane M, Iga N, Nishikawa H, Yamamoto S, Otani S, Waki N, Hirayama S, Miyoshi K, Sugimoto S, Toyooka S, Oto T, Matsukawa A, Miyoshi S (2013). MAPK/ERK pathway activation leads to severe ischemia-reperfusion-induced lung injury. Journal of Heart and Lung Transplantation 32: S138. DOI 10.1016/j.healun.2013.01.309. [Google Scholar] [CrossRef]
Ovechkin AV, Lominadze D, Sedoris KC, Robinson TW, Tyagi SC, Roberts AM (2007). Lung ischemia-reperfusion injury: implications of oxidative stress and platelet-arteriolar wall interactions. Archives of Physiology and Biochemistry 113: 1–12. DOI 10.1080/13813450601118976. [Google Scholar] [CrossRef]
Padro CJ, Sanders VM (2014). Neuroendocrine regulation of inflammation. Seminars in Immunology 26: 357–368. DOI 10.1016/j.smim.2014.01.003. [Google Scholar] [CrossRef]
Perkins GD, Gao F, Thickett DR (2008). In vivo and in vitro effects of salbutamol on alveolar epithelial repair in acute lung injury. Thorax 63: 215–220. DOI 10.1136/thx.2007.080382. [Google Scholar] [CrossRef]
Perkins GD, Mcauley DF, Thickett DR, Gao F (2006). The β-Agonist Lung Injury Trial (BALTI) a randomized placebo-controlled clinical trial. American Journal of Respiratory and Critical Care Medicine 173: 281–287. DOI 10.1164/rccm.200508-1302OC. [Google Scholar] [CrossRef]
Sakamoto J, Chen F, Nakajima D, Yamada T, Ohsumi A, Zhao X, Sakai H, Bando T, Date H (2012). The effect of beta-2 adrenoreceptor agonist inhalation on lungs donated after cardiac death in a canine lung transplantation model. Journal of Heart and Lung Transplantation 31: 773–779. DOI 10.1016/j.healun.2012.03.012. [Google Scholar] [CrossRef]
Sayah DM, Mallavia B, Liu F, Ortiz-Muñoz G, Caudrillier A, DerHovanessian A, Ross DJ, Lynch III JP, Saggar R, Ardehali A, Ware LB, Christie JD, Belperio JA, Looney MR (2015). Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. American Journal of Respiratory and Critical Care Medicine 191: 455–463. DOI 10.1164/rccm.201406-1086OC. [Google Scholar] [CrossRef]
Gao Smith F, Perkins GD, Gates S, Young D, Mcauley DF, Tunnicliffe W, Khan Z, Lamb SE (2012). Effect of intravenous β-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2A multicentre, randomised controlled trial. Lancet 379: 229–235. DOI 10.1016/S0140-6736(11)61623-1. [Google Scholar] [CrossRef]
Takahata Y, Takarada T, Iemata M, Yamamoto T, Nakamura Y et al. (2009). Functional expression of β2 adrenergic receptors responsible for protection against oxidative stress through promotion of glutathione synthesis after Nrf2 upregulation in undifferentiated mesenchymal C3H10T1/2 stem cells. Journal of Cellular Physiology 218: 268–275. DOI 10.1002/jcp.21594. [Google Scholar] [CrossRef]
The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network (2011). Randomized, placebo-controlled clinical trial of an aerosolized β2-agonist for treatment of acute lung injury. American Journal of Respiratory and Critical Care Medicine 184: 561–568. DOI 10.1164/rccm.201012-2090OC. [Google Scholar] [CrossRef]
Uzkeser H, Cadirci E, Halici Z, Odabasoglu F, Polat B, Yuksel TN, Ozaltin S, Atalay F (2012). Anti-inflammatory and antinociceptive effects of salbutamol on acute and chronic models of inflammation in rats: involvement of an antioxidant mechanism. Mediators of Inflammation 2012: 1–10. DOI 10.1155/2012/438912. [Google Scholar] [CrossRef]
Vivona ML, Matthay M, Chabaud MB, Friedlander G, Clerici C (2001). Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: Reversal by β-adrenergic agonist treatment. American Journal of Respiratory Cell and Molecular Biology 25: 554–561. DOI 10.1165/ajrcmb.25.5.4420. [Google Scholar] [CrossRef]
Ware LB, Landeck M, Koyama T, Zhao Z, Singer J, Kern R, Neidlinger N, Nguyen J, Johnson E, Janz DR, Bernard GR, Lee JW, Matthay MA, the California Transplant Donor Network (2014). A randomized trial of the effects of nebulized albuterol on pulmonary edema in brain-dead organ donors. American Journal of Transplantation 14: 621–628. DOI 10.1111/ajt.12564. [Google Scholar] [CrossRef]
Zhang W, Fievez L, Cheu E, Bureau F, Rong W et al. (2010). Anti-inflammatory effects of formoterol and ipratropium bromide against acute cadmium-induced pulmonary inflammation in rats. European Journal of Pharmacology 628: 171–178. DOI 10.1016/j.ejphar.2009.11.015. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |