DOI:10.32604/biocell.2021.015396
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.015396 | www.techscience.com/journal/biocell |
| Article |
Incidence, genomic diversity, and evolution of strawberry mottle virus in China
College of Horticulture, China Agricultural University, Beijing, 100193, China
*Address correspondence to: Hongqing Wang, wanghq@cau.edu.cn
Received: 16 December 2020; Accepted: 18 February 2021
Abstract: Strawberry mottle virus (SMoV) is one of the most common viruses infecting strawberries, causing losses to fruit yield and quality. In this study, 165 strawberry leaf samples were collected from six provinces of China, 46 of which tested positive for SMoV. The complete genome sequences of 11 SMoV isolates were obtained from Liaoning (DGHY3, DGHY16-2, DGHY17, DGHY20-2, DGHY21, DGHY26-2), Shandong (SDHY1, SDHY5, SDHY31-2, SDHY33-2), and Beijing (BJMX7). The RNA1 and RNA2 nucleotide identities between the 11 Chinese isolates were 95.4–99.3% and 96.3–99.6%, respectively, and they shared 78.4–96.6% and 84.8–93.5% identities with the available SMoV isolates in GenBank. Recombination analysis revealed that Chinese isolate SDHY33-2 and Canadian isolates Ontario and Simcoe were recombinants, and recombination events frequently occurred in the 3’ UTR of SMoV. Phylogenetic analysis showed that in an RNA1 tree, most Chinese isolates clustered into the same group while isolate DGHY17 clustered into another group together with Czech isolate C and three Canadian isolates. In an RNA2 tree, all Chinese isolates clustered into a single group. The phylogenetic analysis based on nucleotide sequences was consistent with the results based on coat protein (CP) and RNA-dependent RNA polymerase (RdRp). Further evolutionary analysis indicated that negative selection drives SMoV evolution, and gene flow plays a major role in genetic differentiation. Additionally, reassortment and recombination also influence the evolution of SMoV. To our knowledge, this is the first report of the complete genome of SMoV isolates from China and a detailed analysis of the SMoV population structure.
Keywords: Viral occurrence; Complete genome; Molecular variation; Genetic structure
Strawberry mottle virus (SMoV) was first described as a distinct virus in 1946 (Prentice and Harris, 1946; Martin and Tzanetakis, 2006). It is the most economically important and common virus infecting strawberry (Fragaria spp.) in Europe and North America (Tzanetakis and Martin, 2013; Cieślińska, 2019). The virus is found in all areas where its vectors (Apbis gossypii and Cbaetosipbon species) are present, and it can infect all species of strawberry (Thompson and Jelkmann, 2003). SMoV causes up to 30% losses in fruit yield and runner production, even though most modern cultivars do not exhibit any obvious symptoms upon a single infection (Thompson et al., 2002; Yang et al., 2009; Tzanetakis and Martin, 2013). When co-infecting with other strawberry viruses such as SVBV, SMYEV, and/or SCV, SMoV can reduce the vigor and yield of strawberry plants by up to 80% (Thompson and Jelkmann, 2003). SMoV contains two positive-sense RNA genome segments, and each encodes a polyprotein (Thompson et al., 2002). The RNA1 polyprotein (P1) is cleaved by 3CL-Pro into a putative helicase (Hel), a 3C-like protease (3CL-Pro), a viral genome-linked protein (VPg), and an RNA-dependent RNA polymerase (RdRp) at its C-terminus, and two unknown proteins (X1 and X2) at its N-terminus (Mann et al., 2017). The 3CL-Pro enzyme also cuts the RNA2 polyprotein (P2) at a single site to release the predicted movement protein (MP). The RNA2-encoded glutamic protease then cleaves P2 at two sites to release the putative coat protein (CP), glutamic protease (Pro2Glu), and an unknown protein (Mann et al., 2017; Mann et al., 2019). In 2004 SMoV was reported to be a member of the Sadwavirus genus (Martin and Tzanetakis, 2006; Sanfaçon et al., 2020). However, it was demoted from the genus Sadwavirus in 2009 because the number of CPs was unclear (Sanfaçon et al., 2009). Recently, analysis of cleavage sites in P2 of SMoV revealed that this virus encodes one putative large CP (Mann et al., 2019), indicating that SMoV is not a typical member of the Sadwavirus genus, which possess two CPs. Additionally, SMoV also encodes a novel type of viral glutamic protease that is not present in the Secoviridae family, apart from the black raspberry necrosis virus (BRNV) (Mann et al., 2019; Sanfaçon et al., 2020). Therefore, the International Committee on the Taxonomy of Viruses (ICTV) Secoviridae Study Group proposed to create a subgenus called ‘Stramovirus’ within the genus Sadwavirus. SMoV and BRNV were subsequently classified into the subgenus Stramovirus (Sanfaçon et al., 2020).
Based on data from the Food and Agriculture Organization of the United Nations (FAO), China is the world’s largest producer of strawberries in terms of area harvested. Some viruses, including SMoV, strawberry vein banding virus (SVBV), strawberry mild yellow edge virus (SMYEV), strawberry crinkle virus (SCV) (Wang et al., 1991), cucumber mosaic virus (CMV) (Chen et al., 2014), strawberry necrotic shock virus (SNSV) (Li and Yang, 2011) and various others have been found to infect strawberry in the major production areas of China. Among these viruses, SMoV and SVBV occur frequently in most strawberry production areas of China (Xi, 2017; Wang et al., 2020). In recent years, SMoV was detected by transmission to susceptible indicator plants and reverse transcription-polymerase chain reaction (RT-PCR; Thompson and Jelkmann, 2003), but more convenient, sensitive, and specific detection methods such as real-time quantitative RT-PCR (RT-qPCR) and enzyme-linked immunosorbent assay are unestablished. Up to December 2020, seven complete genome sequences (one from the Netherland and six from Canada) of SMoV are available in the GenBank database; however, the complete genome sequence from China is still unreported, and the evolutionary characteristics between them remain unknown.
Herein, the complete genome sequences of 11 SMoV isolates from China were determined and annotated, and the sequence, recombination, phylogenetic, and population structure analyses were also performed. Our study would lay a foundation for developing molecular diagnosis and effective disease control strategies for this damaging pathogen.
In this study, 165 strawberry leaf samples were collected from the main strawberry production areas of China, including Beijing (68 samples), Anhui (20 samples), Shandong (22 samples), Liaoning (35 samples), Xinjiang (17 samples), and Sichuan (3 samples). The presence of SMoV, SVBV, SMYEV, SCV, CMV, and SNSV was investigated. Total RNA was extracted from leaf tissues using an E.Z.N.A. Plant RNA Kit (Omega Bio-tek, Norcross, USA) following the manufacturer’s instructions. For reverse transcription (RT), total 10 μL of reaction mixture containing 2 μL M-MLV 5× reaction buffer, 2 μL dNTPs (10 mM), 0.5 μL random hexamer primer (10 mM), 0.5 μL oligo dT (18) primer (10 mM), 0.25 μL recombinant RNasin® ribonuclease inhibitor (50 U/μL), 0.25 μL M-MLV Reverse Transcriptase (200 U/μL ; Promega, Madison, USA), 1 μL total RNA (1 μg/μL) and 3.5 μL nuclease-free water was prepared and incubated at 42°C for 1 h. PCR was performed using Taq polymerase (Tiangen, Beijing, China) with specific primers (Tab. S1). The reaction mixture (20 μL) consisted of 10 μL 2× Taq PCR Mix, 0.8 μL sense and antisense primers, respectively, 1 μL cDNA and 7.4 μL nuclease-free water, and then was denatured at 95°C for 3 min and followed by 35 cycles of PCR amplification at 95°C for 30 s, 50°C (SMoV, SVBV and SMYEV; 54°C for SCV; 58°C for SNSV; 56°C for CMV) for 30 s, 72°C for 30 s and a final elongation step of 5 min at 72°C. The 11 SMoV-positive samples (DGHY3, DGHY16-2, DGHY17, DGHY20-2, DGHY21, and DGHY26-2 are ‘Benihope’ cultivars from Donggang in Liaoning province; SDHY1, SDHY5, SDHY31-2, and SDHY33-2 are ‘Benihope’ cultivars from Shandong, and BJMX7 is a ‘Miaoxiang 7’ cultivar from Beijing) were subjected to full-length amplification of the SMoV genome.
For determination of the 5’ and 3’ cDNA ends of the genomic RNA, a SMARTer RACE 5’/3’ Kit (Clontech, California, USA) was used according to the manufacturer’s instructions. For genomic sequences, reverse transcription was performed using a PrimeScript II 1st Strand cDNA Synthesis Kit (TaKaRa, Kusatsu, Japan), and Q5 High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, USA) was subsequently employed for PCR. Three overlapping PCR fragments were amplified for each RNA1 and RNA2 to obtain the SMoV isolates DGHY3, DGHY16-2, DGHY17, DGHY20-2, DGHY26-2, SDHY1, SDHY5, SDHY31-2, SDHY33-2, and BJMX7. The isolate DGHY21 was amplified successfully using primers of 5’ and 3’ terminal sequences. The primers used in this study were listed in Tab. S1. The PCR products were gel-purified (Axygen, Union, USA), cloned into the pTOPO-Blunt vector (Aidlab, Beijing, China), and sequenced by Sangon Biotech (Shanghai) Co., Ltd. The resulting sequences were assembled with default parameters using Seqman within DNASTAR Lasergene v7.1.0 (DNASTAR Inc., Madison, USA).
The complete genome sequences of 11 SMoV Chinese isolates have been deposited in GenBank with the following accession numbers: for SMoV RNA1 sequence, DGHY3, DGHY21, SDHY1, SDHY5, and BJMX7 is MT070747–MT070751, respectively; DGHY16-2, DGHY17, DGHY20-2, DGHY26-2, SDHY31-2, and SDHY33-2 is MT991093–MT991098, respectively. For SMoV RNA2 sequence, DGHY3, DGHY21, SDHY1, SDHY5, and BJMX7 is MT070752–MT070756, respectively; DGHY16-2, DGHY17, DGHY20-2, DGHY26-2, SDHY31-2, and SDHY33-2 is MT991099–MT991104, respectively (https://www.ncbi.nlm.nih.gov/nuccore/?term=strawberry+mottle+virus).
Nucleotide (nt) and deduced amino acid (aa) sequences were aligned using ClustalW in MegAlign of DNASTAR Lasergene v7.1.0. The percentage of homology was then calculated according to the Martinez-NW method using MegAlign v7.1.0. Phylogenetic trees were constructed with MEGA 6.06 software (Tamura et al., 2013) using maximum likelihood (ML; Tamura and Nei, 1993) and neighbor-joining (NJ; Saitou and Nei, 1987) methods with 1000 bootstrap replications (Felsenstein, 1985). SMoV RNA, CP, and RdRp full coding sequences of 11 Chinese isolates and ten (one from the Netherland, six from Canada, and three from the Czech Republic) available isolates in GenBank by December 2020 were analyzed. Recombination events were examined based on SMoV complete genome sequences using a suite of seven prediction programs implemented in the RDP4 software package (Martin et al., 2015). Only events detected by six or more detection methods with default parameters (highest acceptable probability value = 0.05) were considered. DnaSP v5.1 (Librado and Rozas, 2009) was used to estimate Tajima’s D, Fu and Li’s D* and F* statistical tests, nonsynonymous (dN) and synonymous (dS) substitutions, nucleotide and haplotype diversity, genetic differentiation, and gene flow for SMoV coding regions.
The incidence and distribution of strawberry viruses
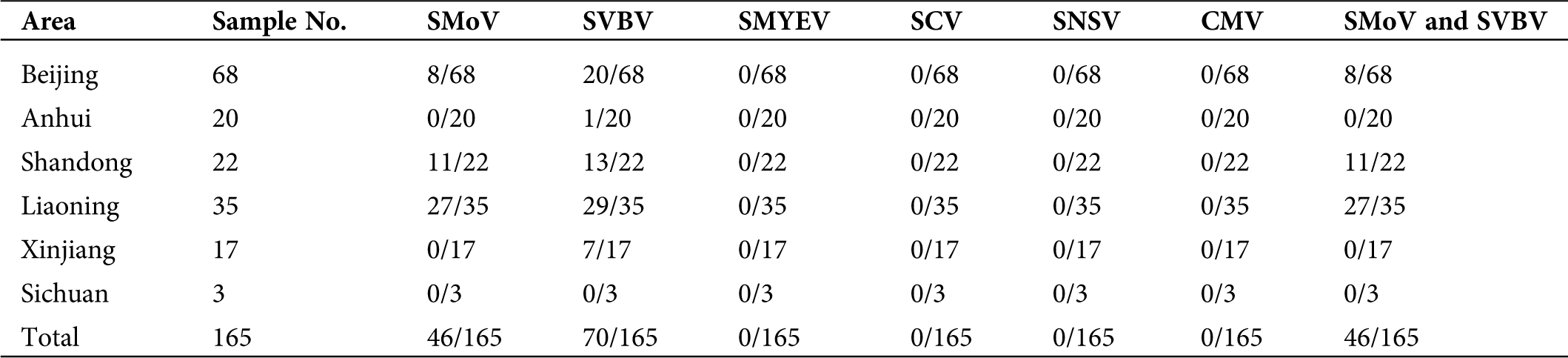
To investigate the incidence and distribution of six viruses infecting strawberry plants, a total of 165 strawberry leaf samples were collected randomly in six provinces of China. Among these samples, 46 (27.9%) were positive for SMoV, 70 (42.4%) for SVBV, while negative for the other four viruses (Tab. S2). All SMoV-positive samples which were collected from Liaoning, Shandong, and Beijing tested positive for SVBV. Liaoning province suffered the worst viral disease rate, with single infection rates for SMoV and SVBV of 77.1% (27/35) and 82.9% (29/35), respectively, and a mixed infection rate of 77.1% (27/35; Tab. S2 and Fig. S1). But no significant correlation was found between the viral presence and symptom. Asymptomatic samples from Beijing (Fig. 1H) and Liaoning (Fig. 1J) showed SMoV and SVBV positive while symptomatic samples with mottled (Figs. 1D, 1E, 1G and 1L), distorted (Figs. 1A and 1D), crinkled (Figs. 1C and 1E), deformed (Fig. 1G) and/or purplish red (Fig. 1K) on the leaves were negative for the six tested viruses.

Figure 1: The pictures of representative samples, collected from different regions of China.
Genomic characteristics of the Chinese SMoV isolates
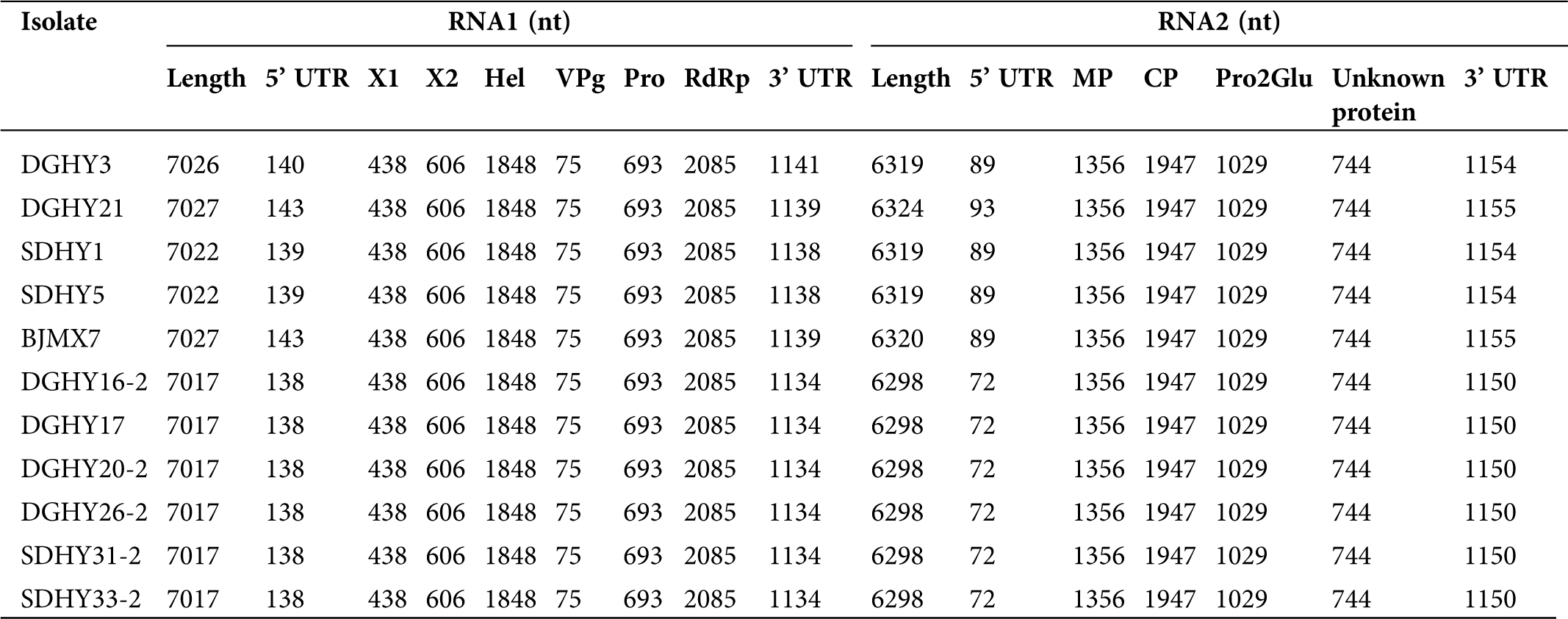
The genomic RNA1 sequences of the 11 Chinese isolates ranged from 7017 to 7027 nt, excluding the poly (A) tail (Tab. S3). All of these 11 isolates encoded polyprotein P1. Previously identified P1 cleavage sites (Q146/G, Q348/S, Q964/G, Q989/G, and Q1220/G) were conserved in all Chinese isolates. Subsequent pair-wise comparisons of the RNA1 and polyprotein sequences were performed, and the nt and aa identities were 95.4–99.3% and 98.1–99.7%, respectively, among the 11 Chinese isolates. Collectively, the 11 Chinese isolates shared the highest sequence identities (95.5–96.6% nt and 98.3–99.3% aa) with Canadian isolate NSper3 and the lowest sequence identities (78.4–79.5% nt and 88.3–89.2% aa) with Netherlandish isolate 1134.
The genomic RNA2 sequences of the 11 Chinese isolates ranged from 6298 to 6324 nt, excluding the poly (A) tail (Tab. S3). These 11 isolates all encoded polyprotein P2. Previously identified P2 cleavage sites (E452/G, P1101/AFP, and P1444/KFP) were conserved in all Chinese isolates. Pair-wise comparisons of the 11 Chinese isolates revealed nt and aa identifies of 96.3–99.6% and 97.7–99.9%, respectively. Additionally, the 11 Chinese isolates shared similar sequence identities (92.6–93.5% nt and 96.9–97.7% aa) with NB926, Ontario, and NSper17 isolates from Canada and isolate 1134 from the Netherlands (92.6–93.1% nt and 96.2–96.8% aa). The lowest sequence identities were shared with Canadian isolate NSper3 (84.8–85.2% nt and 94.4–94.7% aa). Overall, the genomic RNA1 region of the 11 Chinese isolates shared a higher sequence identity with Canadian isolates than Netherlandish isolate 1134, but there were no obvious geographic differences between RNA2 sequences.
Phylogenetic trees were constructed based on SMoV RNA1, RNA2, CP, and RdRp full coding regions using the ML and NJ methods. Only ML trees are shown in this study because both ML and NJ trees displayed almost identical topologies. In the RNA1 tree, most Chinese isolates clustered into the same group while DGHY17 clustered into another group together with Czech isolate C and Canadian isolate NSper3, NSper17, and NB926, and shared a close relationship with Czech C. The Netherlandish isolate 1134 and the Canadian isolate NSper51 and Simcoe formed a different branch furthest from all Chinese isolates. Czech isolates A and B formed two separate groups, respectively (Fig. 2A). Similar results were obtained when the analysis was conducted with the RdRp coding region within RNA1 (Fig. S2A). These results indicated that Czech and Canadian isolates showed greater molecular variation than Chinese isolates.
In the RNA2 tree, all of the 11 Chinese isolates formed a single group, characterized by the close relationship with Czech isolate C and the furthest distance from Czech isolate A. Canadian isolate NB926 and NSper17 were grouped into another branch with Netherlandish isolate 1134. Meanwhile, Canadian isolates NSper51 and NSper3 formed a single group. Czech isolate B also formed a separate group (Fig. 2B). In the CP tree based on the full CP coding region within RNA2, isolates from China, Canada, the Netherlands, and the Czech Republic displayed phylogenetic relationships that were consistent with those based on the RNA2 whole coding sequences (Fig. S2B). Concurrently, phylogenetic analysis revealed no obvious tendency for isolates to group according to geographical origin among different countries, but a clear tendency among Liaoning, Shandong, and Beijing of China.

Figure 2: Phylogenetic analysis of strawberry mottle virus (SMoV) Chinese isolates and available isolates from GenBank based on whole coding sequences of RNA1 (A) and RNA2 (B) using the maximum likelihood (ML) method with 1000 bootstrap replicates.
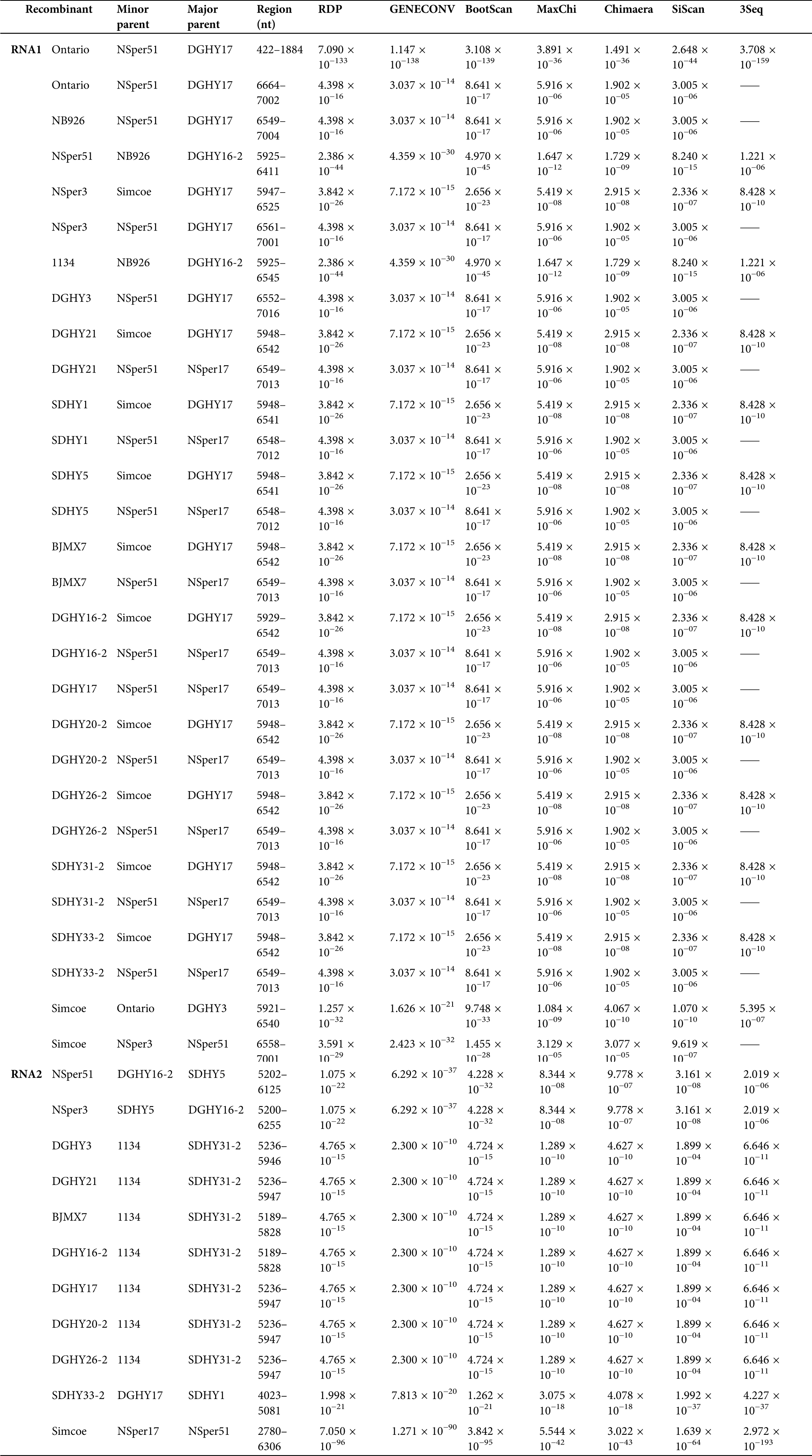
A recombination event was detected by all seven methods with p-values ranging from 3.708 × 10−159 to 3.891 × 10−36, and the recombination junction located at the RNA1 422–1884 nt region of the Ontario isolate (Tab. S4). We also identified two recombination events that were supported with a high degree of confidence in the RNA2 of the SDHY33-2 isolate (4023–5081 nt) and the Simcoe isolate (2780–6306 nt), respectively (Tab. S4). Additionally, in the 3’ UTR of SMoV RNA1 and RNA2, most SMoV isolates were detected recombination events with a moderate degree of confidence (Tab. S4). These results suggest that recombination events frequently occurred in the 3’ UTR of SMoV.
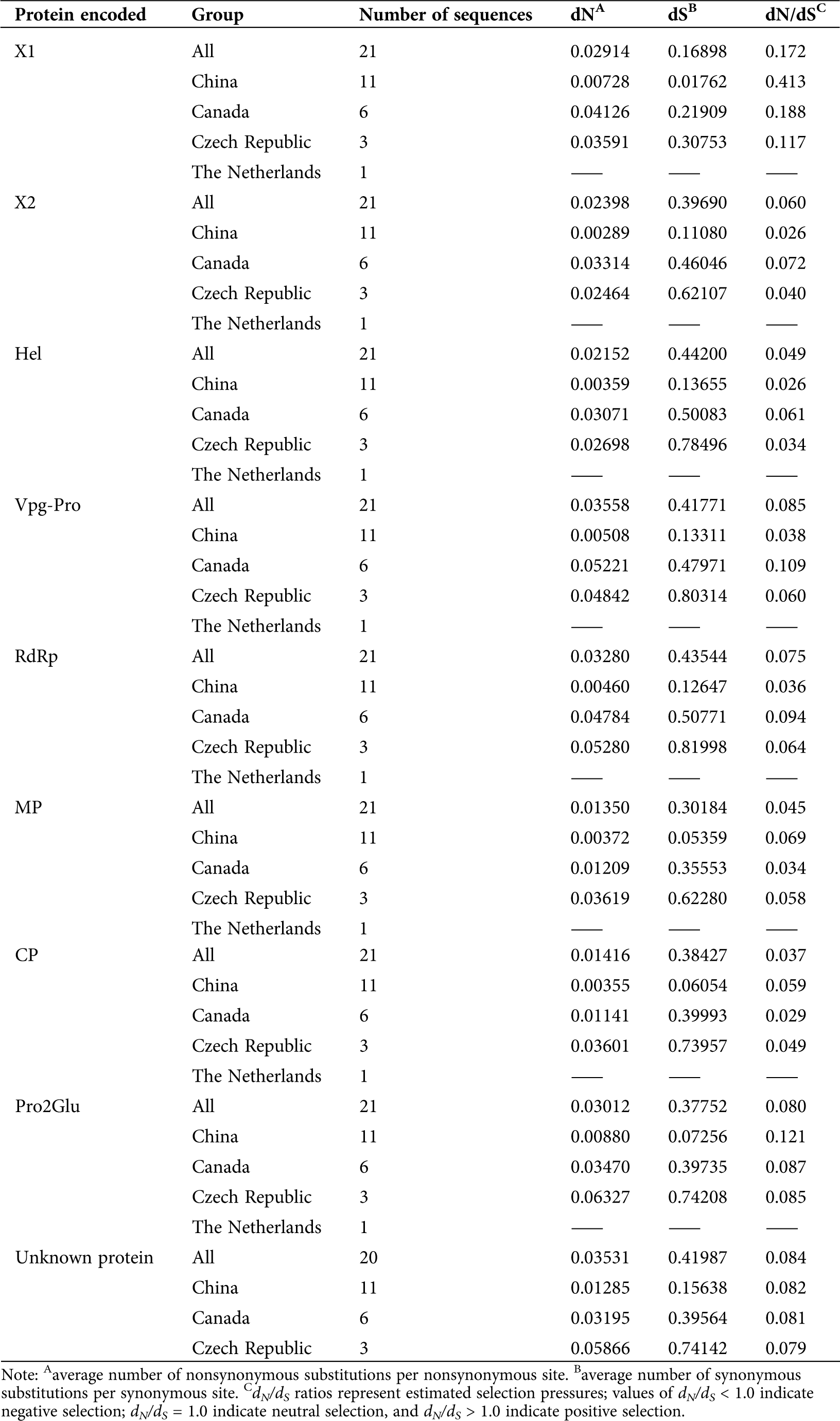
Evolutionary analysis, including selection pressure, neutrality tests, population demography, genetic differentiation, and gene flow, were carried out on the SMoV RNA1 coding region (including X1, X2, Hel, Vpg, 3CL-Pro, and RdRp coding regions) and the RNA2 coding region (including MP, CP, Pro2Glu, and unknown protein-coding regions) based on geographic populations. For selection pressure, the ratios of non-synonymous (dN) and synonymous (dS) sites were calculated. The results revealed that the dN/dS ratios for each protein-coding region of SMoV isolates from China, Canada, and the Czech Republic were <1, suggesting that SMoV populations were under negative selection (Tab. 1).
Table 1: Selection pressure analysis of SMoV protein coding regions based on geographical population
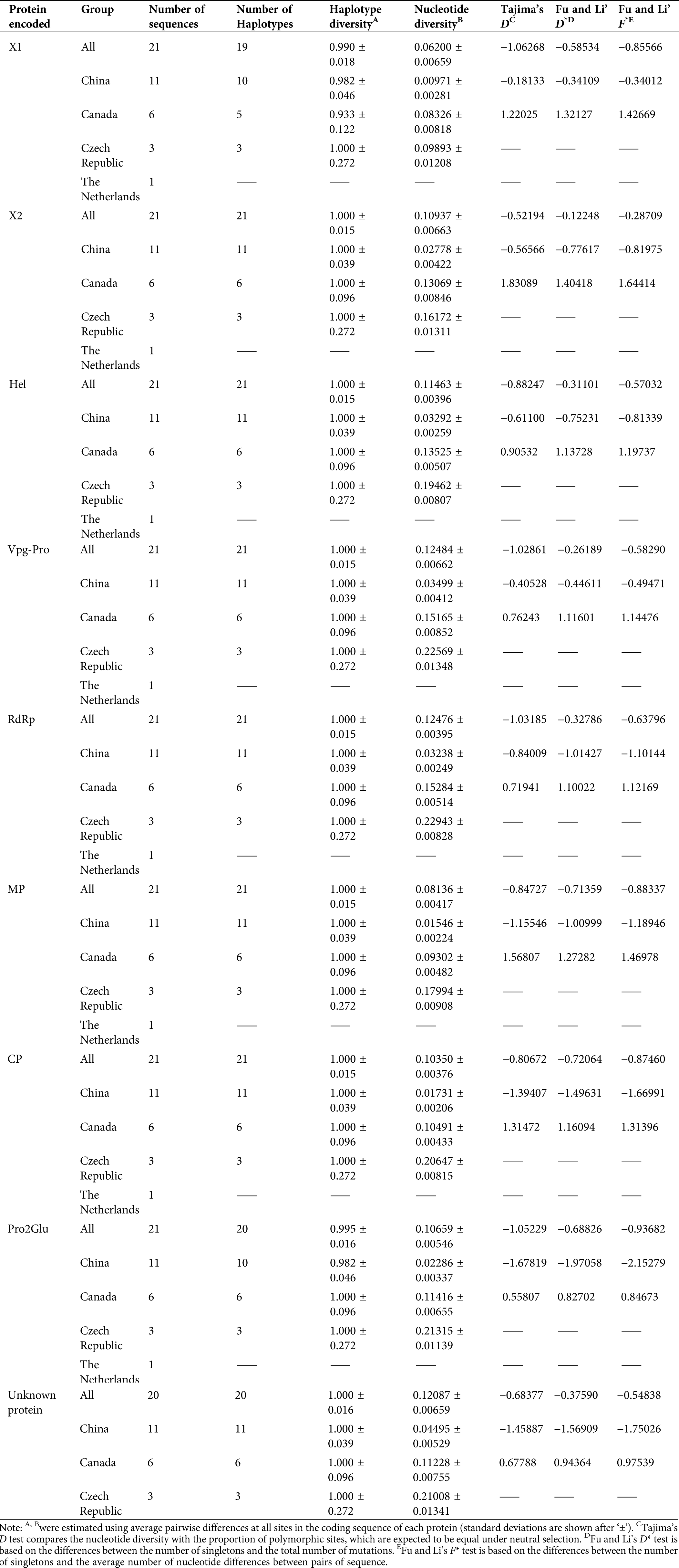
For neutrality tests and population demography, we evaluated the values of Tajima’s D, Fu and Li’s D* and Fu, and Li’s F* statistical tests, as well as haplotype and nucleotide diversities. Except for Canadian isolates, all protein-coding regions from different countries were negative, indicating that most SMoV populations were in a state of expansion. However, their p-values were not significant (Tab. 2). Haplotype and nucleotide diversities for all coding regions were estimated, and high haplotype diversity and low nucleotide diversity were presented for each coding region within individual geographic groups (Tab. 2).
Table 2: Haplotype diversity, nucleotide diversity, and neutrality testing of SMoV protein coding regions based on geographical population
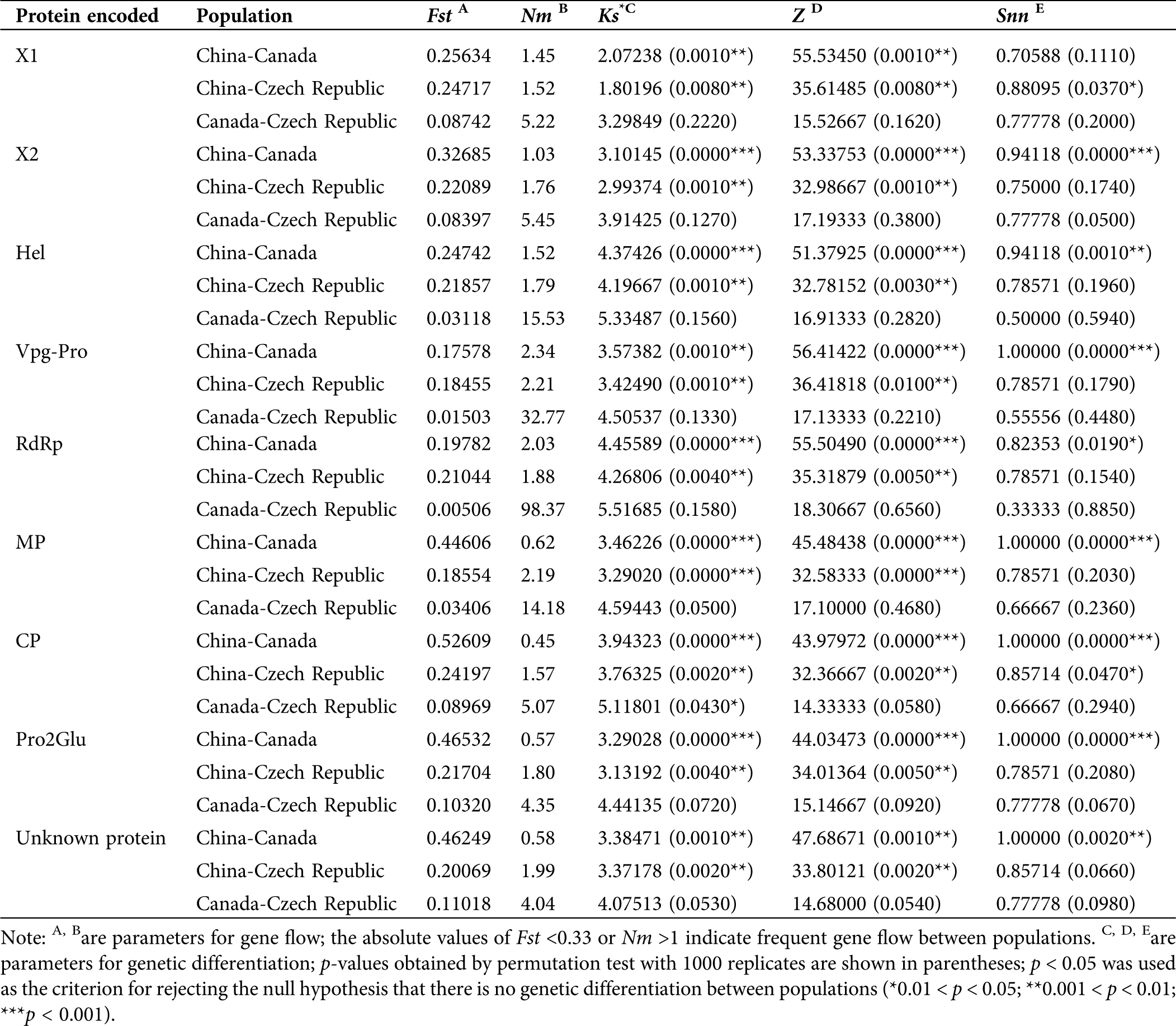
For genetic differentiation, the p-values of Ks*, Z, and Snn were calculated. The P-Ks* and Z values for each protein-coding region between populations from China and populations from Canada and the Czech Republic were between 0.001 and 0.01, and <0.001, respectively, indicating significant genetic differentiation between them. The Ks* and Z values for all coding regions between Canadian and Czech populations were not significantly different, indicating no significant genetic differentiation. Regarding Snn values, most coding regions were significantly different between Chinese and Canadian populations, but other geographical groups showed no significant differences (Tab. 3).
Regarding gene flow, the absolute values of Fst for most coding regions between Chinese, Canadian, and Czech populations were <0.33 (Tab. 3), suggesting frequent gene flow between these populations. The corresponding Nm absolute values for these populations were >1, also indicating that there were pathways for gene flow between them. However, the absolute values of Fst were >0.33 and Nm <1 between Chinese and Canadian RNA2 coding regions (Tab. 3), suggesting infrequent gene flow, hence genetic drift might be the main factor shaping genetic differentiation in RNA2 between Chinese and Canadian populations. Furthermore, the most frequent gene flow occurred between Canadian and Czech populations, based on the lowest Fst and highest Nm absolute values (Tab. 3).
Table 3: Gene flow and genetic differentiation of SMoV protein coding regions based on geographical population
In recent years, the detection rates reported for SMoV, SVBV, SMYEV, SCV, and CMV were 18.9%, 21.7%, 4.2%, 37.3%, and 21%, respectively (Xi, 2017; Wang et al., 2020), in major strawberry production areas of China. SNSV has only been reported in Heilongjiang province. In the present study, only SMoV (27.9%) and SVBV (42.4%) tested positive, and SMoV was detected in three provinces (Liaoning, Shandong, and Beijing) while SVBV was also detected in another two provinces (Xinjiang and Anhui), indicating higher SVBV and SMoV infection rates than reported in the previous studies (Xi, 2017; Wang et al., 2020) and SVBV was widespread in China. The samples from Sichuan were negative for six viruses, which may be because of too few samples. The symptoms of SVBV may be masked in combination with SMoV or by high levels of nitrogen (Martin and Tzanetakis, 2006). Symptomless strawberry samples from Liaoning province exhibited the highest virus infection rates among the six provinces; some symptomatic samples were negative for viruses, suggesting that it is more difficult to identify viral plants by apparent symptoms. Thus, molecular diagnosis is still indispensable for strawberry viruses. Notably, strawberry plants with SVBV and SMoV were more susceptible to strawberry fusarium wilt, powdery mildew, Botrytis cinerea, and red spider than virus-free plants (data not shown). Therefore, developing effective molecular diagnostic technology for SMoV and SVBV remains urgent. The complete genome sequence of SMoV Chinese isolates will be conducive to designing newly specific primers and probes for RT-qPCR and preparing antibodies against SMoV.
The research about the SMoV genome sequence was relatively slow. Only one complete genome sequence (isolate 1134 from the Netherlands) was obtained by 2002 (Thompson et al., 2002). In 2016, five complete genome sequences of SMoV from Canada were reported (Bhagwat et al., 2016), but none have yet been reported from China. In our present study, we obtained the complete genome sequences of 11 Chinese isolates. All Chinese isolates shared high sequence identity and clustered in the same clade in the RNA2 tree, but the DGHY17 isolate was grouped into the same branch with Czech isolate C and three Canadian isolates in the RNA1 tree, suggesting that recombination or reassortment events occurred during the evolution of SMoV isolates. This was also reported previously for Canadian isolate NSper3 (Bhagwat et al., 2016). Further recombination analysis was performed, but no recombination event was identified in the coding sequences of the DGHY17 isolate, indicating that reassortment occurred in the DGHY17 isolate. Additionally, recombination events occurred frequently in the 3′UTRs of both RNA1 and RNA2, suggesting that recombination is an important driving force during the evolution of SMoV. The phylogenetic results reveal that the Chinese isolates kept low molecular variation, but Czech and Canadian isolates happened high molecular variation. This may be related to vectors and strawberry transplants. Recent research demonstrated that SMoV can be transmitted by Chaetosiphon fragaefolii. C. fragaefolii is the most important vectors of viruses in strawberry fields and presumed to originate from North America (Converse, 1987; Fránová et al., 2019). C. fragaefolii has also been found in the South Bohemia area of the Czech Republic (Fránová et al., 2019). Thus, we believe that C. fragaefolii may make a significant contribution to the high variation in the Czech SMoV isolate. In addition, some strawberry transplants used in Canadian production are from the USA (Bonneau et al., 2019), which may assist in virus spreading.
During the evolution of plant viruses, genetic selection and drift are the two main processes (Garcia-Arenal et al., 2003). Previous studies reported that negative selection operates on most animal and plant viruses (Garcia-Arenal et al., 2001; Yin et al., 2013; He et al., 2013). In our current study, SMoV protein populations from China, Canada, and the Czech Republic were subjected to negative selection, indicating strong purifying selection against SMoV mutation as a driving force for SMoV evolution. Among these proteins, the dN/dS ratios for X1 were much greater than for other proteins, indicating that constraints on X1 were higher than for other proteins; hence, the X1 sequence may be more highly conserved.
Genetic differentiation and gene flow analysis revealed no genetic differentiation between Canadian and Czech SMoV populations, and consistent with this observation, the most frequent gene flow was found between these groups. This may be due to the expansion of the C. fragaefolii vector from Canada to the Czech Republic. According to previous studies (Fránová et al., 2019; CABI, 2019), the C. fragaefolii may originate from North America and spread widely in Europe. Strawberry or other plant material imports and exports may assist the C. fragaefolii in spreading by carrying them. There was no gene flow between Chinese RNA2 and Canadian RNA2 populations, and genetic differentiation was significant. Interestingly, there was genetic differentiation between Chinese and Czech SMoV populations, although gene flow was identified between them. These results indicate that gene flow was the main element influencing SMoV genetic differentiation, but some other factors such as gene drift, recombination, or reassortment may also drive SMoV evolution.
In conclusion, our study provides the first complete genome sequences of SMoV isolates from China. The Chinese isolates shared high sequence identity, but Czech isolates occurred high molecular variation and existed frequent gene flow with Canadian isolates. Negative selection drove SMoV population evolution, and gene flow played a major role in SMoV genetic differentiation. In addition, reassortment and recombination also influence the structure of SMoV populations. To our knowledge, this is the first detailed analysis of SMoV population structure.
Acknowledgement: We thank Professors Shifang Li and Zhixiang Zhang from the State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, for valuable comments on this study.
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author Contribution: The authors confirm contribution to the paper as follows: study conception and design: Lingjiao Fan, Hongqing Wang; data collection: Lingjiao Fan, Chengyong He, Mengmeng Wu, Dehang Gao, Zhenfei Dong, Shengfan Hou, Zekun Feng; analysis and interpretation of results: Lingjiao Fan; draft manuscript preparation: Lingjiao Fan. All authors reviewed the results and approved the final version of the manuscript.
Ethics Approval: This work did not involve any studies on human participants or animals.
Funding Statement: This study was funded by the National Key R&D Program of China (2019YFD1001800).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Bhagwat B, Dickison V, Ding XL, Walker M, Bernardy M, Bouthillier M, Creelman A, Young RD, Li YZ, Nie XZ, Wang AM, Xiang Y, Sanfaçon H (2016). Genome sequence analysis of five Canadian isolates of strawberry mottle virus reveals extensive intra-species diversity and a longer RNA2 with increased coding capacity compared to a previously characterized European isolate. Archives of Virology 161: 1657–1663. DOI 10.1007/s00705-016-2799-6. [Google Scholar] [CrossRef]
Bonneau P, Hogue R, Tellier S, Fournier V (2019). Evaluation of various sources of viral infection in strawberry fields of Quebec. Canada Journal of Economic Entomology 112: 2577–25837. DOI 10.1093/jee/toz205. [Google Scholar] [CrossRef]
Cieślińska M (2019). Genetic diversity of seven Strawberry mottle virus isolates in Poland. Plant Pathology Journal 35: 389–392. DOI 10.5423/PPJ.NT.12.2018.0306. [Google Scholar] [CrossRef]
CABI (2019). Plantwise Knowledge Bank, Chaetosiphon fragaefolii. https://www.plantwise.org/knowledgebank/datasheet/13305#DistributionSection. [Google Scholar]
Converse RH (1987). Virus disease of small fruits. In: Agricultural Handbook. Faculty Publications in the Biological Sciences 631: xii, 277. USDA: ARS. [Google Scholar]
Chen L, Shang QX, Chen XY, Xing DM, Yang R, Ran C, Wei YM, Zhao XY, Liu ZP (2014). First report on the occurrence of Cucumber mosaic virus on Fragaria ananassa in China. Plant Disease 98: 1015. DOI 10.1094/PDIS-11-13-1173-PDN. [Google Scholar] [CrossRef]
Felsenstein J (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39: 783–791. DOI 10.1111/j.1558-5646.1985.tb00420.x. [Google Scholar] [CrossRef]
Fránová J, Přibylová J, Koloniuk I (2019). Molecular and biological characterization of a new strawberry cytorhabdovirus. Viruses 11: 982. DOI 10.3390/v11110982. [Google Scholar] [CrossRef]
Garcia-Arenal F, Fraile A, Malpica JM (2003). Variation and evolution of plant virus populations. International Microbiology 6: 225–232. DOI 10.1007/s10123-003-0142-z. [Google Scholar] [CrossRef]
Garcia-Arenal F, Fraile A, Malpica JM (2001). Variability and genetic structure of plant virus populations. Annual Review of Phytopathology 39: 157–186. DOI 10.1146/annurev.phyto.39.1.157. [Google Scholar] [CrossRef]
He Z, Li WF, Yasaka R, Huang YK, Zhang ZX, Ohshima K, Li SF (2013). Molecular variability of sugarcane streak mosaic virus in China based on an analysis of the P1 and CP protein coding regions. Archives of Virology 159: 1149–1154. DOI 10.1007/s00705-013-1854-9. [Google Scholar] [CrossRef]
Librado P, Rozas J (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. DOI 10.1093/bioinformatics/btp187. [Google Scholar] [CrossRef]
Li L, Yang H (2011). First report of Strawberry necrotic shock virus in China. Plant Disease 95: 1198. DOI 10.1094/PDIS-02-11-0121. [Google Scholar] [CrossRef]
Mann KS, Chisholm J, Sanfaçon H (2019). Strawberry mottle virus (family Secoviridae, order Picornavirales) encodes a novel glutamic protease to process the RNA2 polyprotein at two cleavage sites. Journal of Virology 93: e01679-18. [Google Scholar]
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution 1: vev003. DOI 10.1093/ve/vev003. [Google Scholar] [CrossRef]
Martin RR, Tzanetakis IE (2006). Characterization and recent advances in detection of strawberry viruses. Plant Disease 90: 384–396. DOI 10.1094/PD-90-0384. [Google Scholar] [CrossRef]
Mann KS, Walker M, Sanfaçon H (2017). Identification of cleavage sites recognized by the 3C-Like cysteine protease within the two polyproteins of strawberry mottle virus. Frontiers in Microbiology 8: 745. DOI 10.3389/fmicb.2017.00745. [Google Scholar] [CrossRef]
Prentice IW, Harris RV (1946). Resolution of strawberry virus complexes by means of the aphid vector Capitophorus fragariae Theb. Annals of Applied Biology 33: 50–53. DOI 10.1111/j.1744-7348.1946.tb06273.x. [Google Scholar] [CrossRef]
Sanfaçon H, Dasgupta I, Fuchs M, Karasev AV, Petrzik K, Thompson JR, Tzanetakis I, Vlugt R, Wetzel T, Yoshikawa N (2020). Proposed revision of the family Secoviridae taxonomy to create three subgenera, Satsumavirus, Stramovirus and Cholivirus, in the genus Sadwavirus. Archives of Virology 165: 527–533. DOI 10.1007/s00705-019-04468-7. [Google Scholar] [CrossRef]
Saitou N, Nei M (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4: 406–425. [Google Scholar]
Sanfaçon H, Wellink J, Le GO, Karasev A, Vlugt R, Wetzel T (2009). Secoviridae: A proposed family of plant viruses within the order Picornavirales that combines the families Sequiviridae and Comoviridae, the unassigned genera Cheravirus and Sadwavirus and the proposed genus Torradovirus. Archives of Virology 154: 899–907. DOI 10.1007/s00705-009-0367-z. [Google Scholar] [CrossRef]
Thompson JR, Jelkmann W (2003). The detection and variation of strawberry mottle virus. Plant Disease 87: 385–390. DOI 10.1094/PDIS.2003.87.4.385. [Google Scholar] [CrossRef]
Thompson JR, Leone G, Lindner JL, Jelkmann W, Schoen CD (2002). Characterization and complete nucleotide sequence of Strawberry mottle virus: A tentative member of a new family of bipartite plant picorna-like viruses. Journal of General Virology 83: 229–239. DOI 10.1099/0022-1317-83-1-229. [Google Scholar] [CrossRef]
Tzanetakis IE, Martin RR (2013). Expanding field of strawberry viruses which are important in North America. International Journal of Fruit Science 13: 184–195. DOI 10.1080/15538362.2012.698164. [Google Scholar] [CrossRef]
Tamura K, Nei M (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular Biology and Evolution 10: 512–526. [Google Scholar]
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30: 2725–2729. DOI 10.1093/molbev/mst197. [Google Scholar] [CrossRef]
Wang GP, Liu FC, Guo JX (1991). Identification on strawberry viruses in China. Acta Phytopathologica Sinica 21: 9–14, (In Chinese). [Google Scholar]
Wang J, Zhou Y, Zhu N, Chen L, Ren JD, Yang AZ, Han CG, Shang QX (2020). Detection and analysis of cucumber mosaic virus in strawberry seedlings in some provinces of China. Acta Phytopathologica Sinica 50: 649-656, (In Chinese). https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CAPJ&dbname=CAPJLAST&filename=ZWBL20200407003&v=QjsSmhHqCwhGh1pWrKY0oCIx5Z4kpMiW6gK0zcIJa1RsDtuMp1rj51uKYP26fLkG. [Google Scholar]
Xi X (2017). Application of rapid detection of important strawberry viruses in Beijing. pp. 25. Beijing, China Agricultural University, (In Chinese). [Google Scholar]
Yang HY, Li LL, Dai HY, Zhang ZH (2009). Study on the molecular variation and PCR detection of Strawberry mottle virus. Agricultural Sciences in China 8: 1203–1209. DOI 10.1016/S1671-2927(08)60330-2. [Google Scholar] [CrossRef]
Yin X, Zheng FQ, Tang W, Zhu QQ, Li XD, Zhang GM, Liu HT, Liu BS (2013). Genetic structure of rice black-streaked dwarf virus populations in China. Archives of Virology 158: 2505–2515. DOI 10.1007/s00705-013-1766-8. [Google Scholar] [CrossRef]
Supplementary Materials
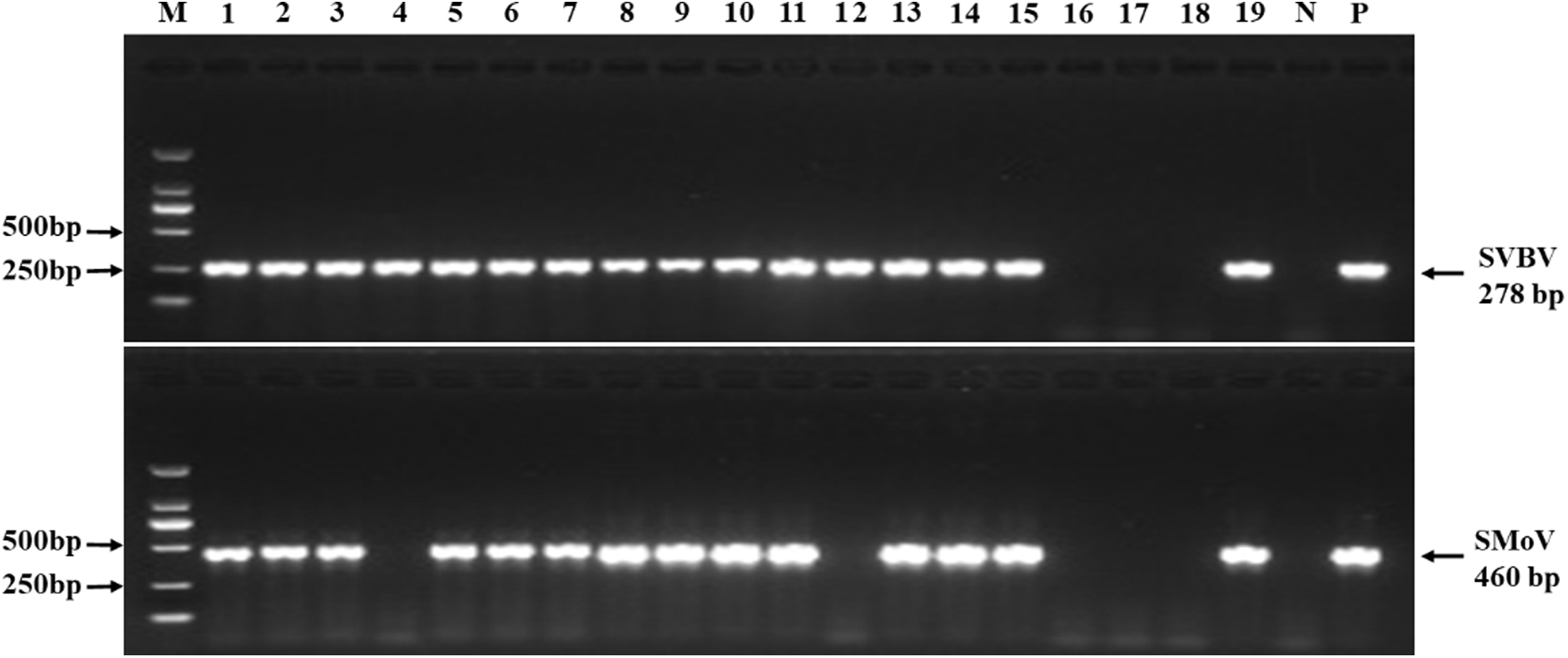
SUPPLEMENTARY Figure 1: Incidence of strawberry vein banding virus (SVBV) and strawberry mottle virus (SMoV) in the strawberry samples from Liaoning of China by RT-PCR.
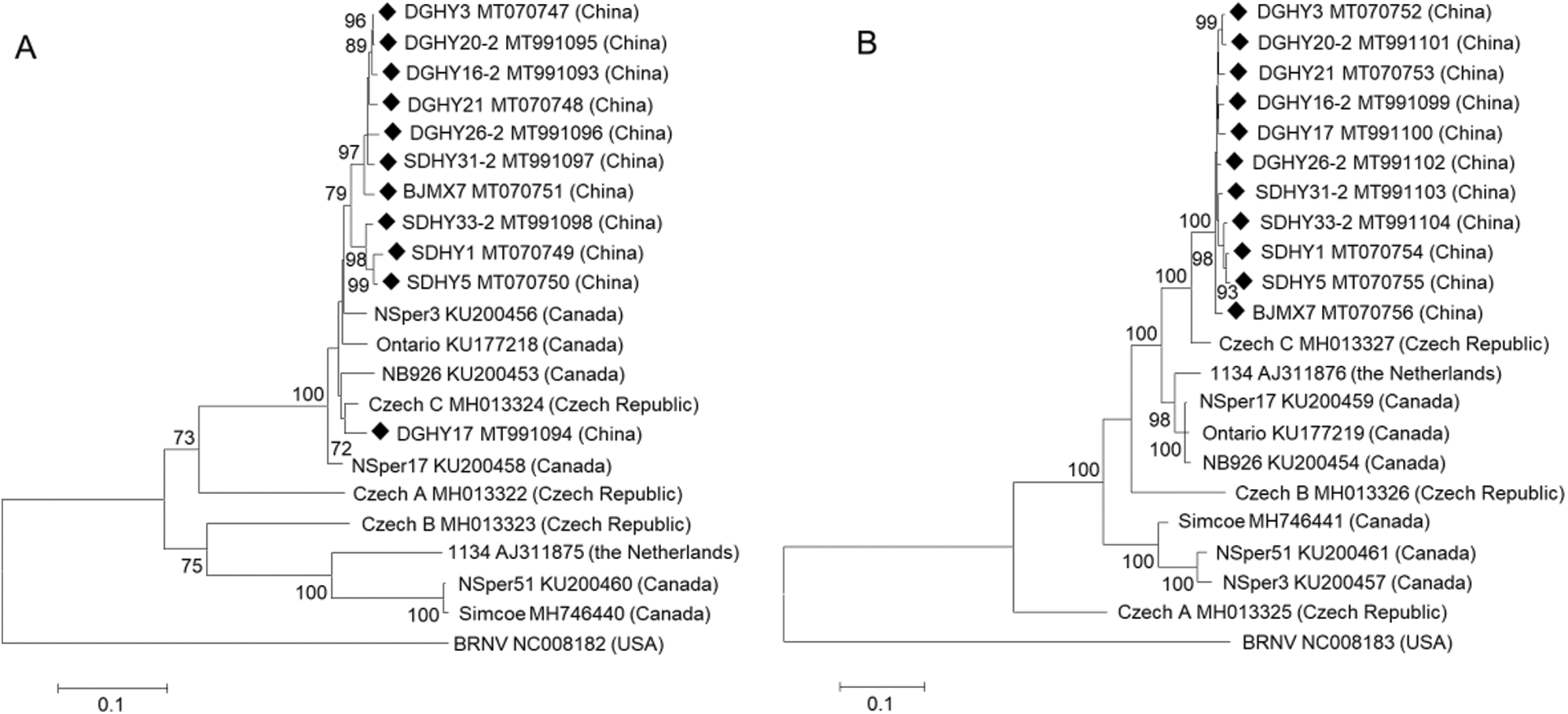
SUPPLEMENTARY Figure 2: Phylogenetic analysis of SMoV Chinese isolates and available isolates from GenBank based on RNA-dependent RNA polymerase (A) and coat protein (B) full coding regions using the ML method with 1000 bootstrap replicates. Only values above 70% are shown.
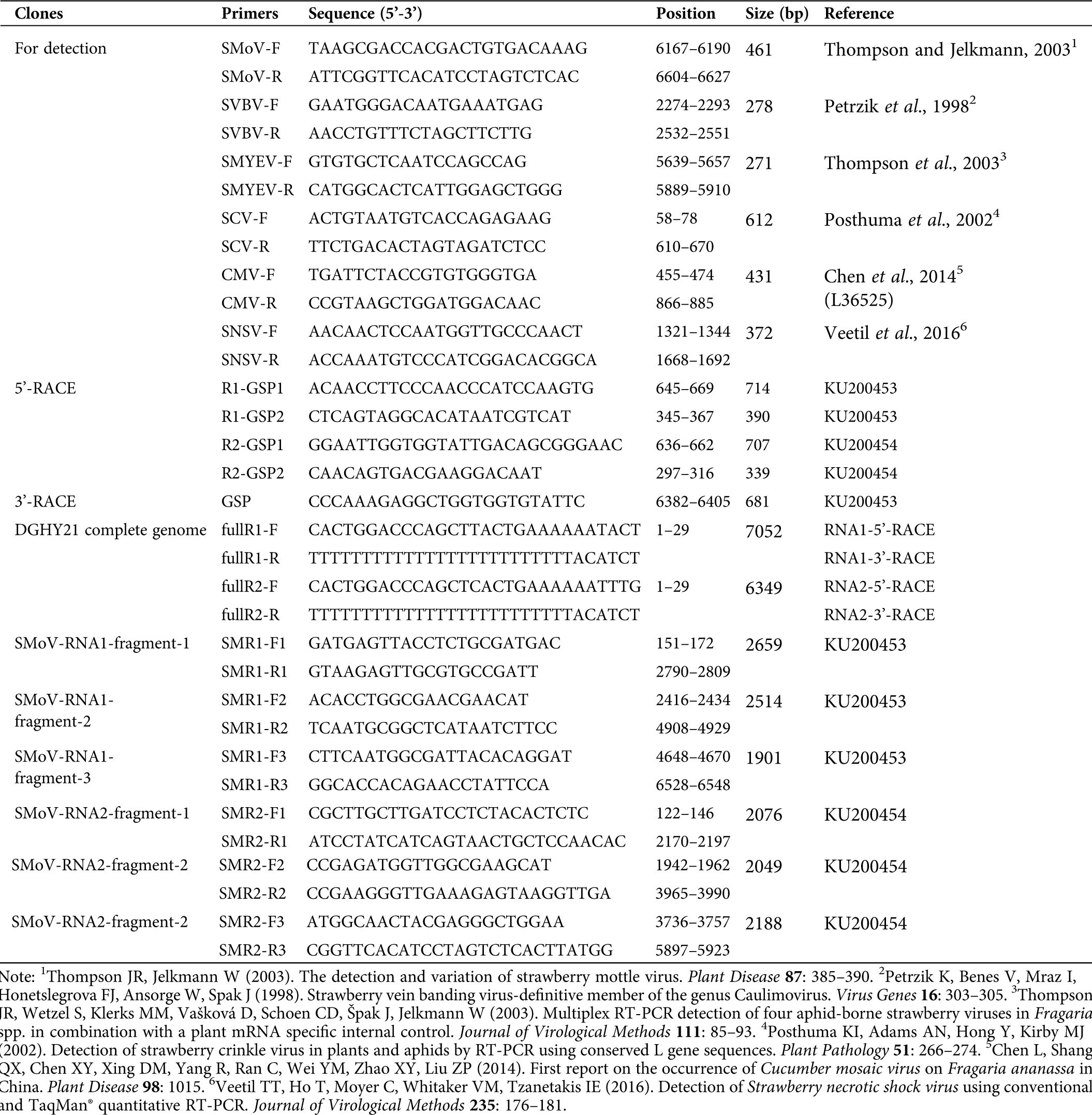
Supplementary Table 1: Primers used in this study
Supplementary Table 2: Detection of viruses in the main strawberry production areas of China
Supplementary Table 3: Genomic characteristics of eleven strawberry mottle virus (SMoV) isolates from China
Supplementary Table 4: Prediction of recombination events
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |