DOI:10.32604/biocell.2021.012938
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.012938 | www.techscience.com/journal/biocell |
| Article |
Integrated analysis of human influenza A (H1N1) virus infection-related genes to construct a suitable diagnostic model
State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, 310003, China
*Address correspondence to: Hongyan Diao, diaohy@zju.edu.cn
Received: 19 July 2020; Accepted: 26 November 2020
Abstract: The genome characteristics and structural functions of coding proteins correlate with the genetic diversity of the H1N1 virus, which aids in the understanding of its underlying pathogenic mechanism. In this study, analyses of the characteristic of the H1N1 virus infection-related genes, their biological functions, and infection-related reversal drugs were performed. Additionally, we used multi-dimensional bioinformatics analysis to identify the key genes and then used these to construct a diagnostic model for the H1N1 virus infection. There was a total of 169 differently expressed genes in the samples between 21 h before infection and 77 h after infection. They were used during the protein–protein interaction (PPI) analysis, and we obtained a total of 1725 interacting genes. Then, we performed a weighted gene co-expression network analysis (WGCNA) on these genes, and we identified three modules that showed significant potential for the diagnosis of the H1N1 virus infection. These modules contained 60 genes, and they were used to construct this diagnostic model, which showed an effective prediction value. Besides, these 60 genes were involved in the biological functions of this infectious virus, like the cellular response to type I interferon and in the negative regulation of the viral life cycle. However, 20 genes showed an upregulated expression as the infection progressed. Other 36 upregulated genes were used to examine the relationship between genes, human influenza A virus, and infection-related reversal drugs. This study revealed numerous important reversal drug molecules on the H1N1 virus. They included rimantadine, interferons, and shikimic acid. Our study provided a novel method to analyze the characteristic of different genes and explore their corresponding biological function during the infection caused by the H1N1 virus. This diagnostic model, which comprises 60 genes, shows that a significant predictive value can be the potential biomarker for the diagnosis of the H1N1 virus infection.
Keywords: Human influenza A; H1N1 virus; Gene; Diagnosis model
Influenza is a highly contagious acute respiratory disease caused by infection of the host respiratory tract that is triggered by influenza viruses. It is characterized by strong and fast infectivity, general susceptibility, high incidence, and is prone to mutational changes (Uyeki et al., 2019). Globally, it has caused several explosive epidemics making it a public health problem of gravest concern (Fineberg, 2014). A novel swine-origin Influenza A (H1N1) virus, one of the most common infectious viruses in humans, is classified within the myxoviridae family of negative-stranded RNA viruses (Rewar et al., 2015). This human influenza could be traced back to the 1918 ‘Spanish flu,’ which approximately infected 500 million people. Since then, in birds and mammals, this virus has led to multiple outbreaks or widespread human influenza (Lyons and Lauring, 2018). A prime example was observed in 2009, and that global pandemic dealt a heavy blow to the public health care sector (Sun et al., 2016). Therefore, to tackle this infectious disease, it is imperative to add research on this existing basic theory by using molecular techniques that seek effective measures to prevent and control this infectious virus.
The seasonal epidemics and occasional pandemics caused by the H1N1 influenza virus are due to its rapid genetic mutation (Zeldovich et al., 2015). During the interaction process between the mutating influenza virus and the human immune barrier, the accumulation of several genetic mutations not only attained immune escape but also weakened the efficacy of the human seasonal influenza vaccine (White and Lowen, 2018). Hence, it is necessary to study the characteristics of the gene mutations of influenza viruses and understand their underlying pathogenic mechanism. Besides, during the infection phase which ranged from mild to more severe pneumonia, the dynamic gene expression changes and complex interactions between different genes present in the developmental stage of the disease lead to significant pathophysiological changes. Hence, understanding the gene dynamics and related functional changes caused by the H1N1 virus infection is critical to elucidate the underlying pathogenesis of this virus and to improve its clinical diagnosis and treatment (Josset et al., 2012). Previous genetic studies showed that F35L gene mutations in the polymerase acidic protein fragment of the H1N1 virus played a significant role in the emergence of any pandemic. Consequently, the polymerase acidic protein fragment was considered as a possible target for the treatment of Influenza A viruses. This was because of its essential role in the transcription and replication processes in viruses (Lutz Iv et al., 2020). Using the genome-wide association study (GWAS) method, Cheng et al. (2015) discovered that high expression of the TMPRSS2 gene found in humans increased the severity of illness associated with influenza virus infection; whereas, mutations in the MPRSS2 gene also increased the susceptibility of human infection with H7N9 avian influenza virus. However, the mechanisms by which gene mutations affect the infectious ability of the H1N1 virus, how the host immune system adapts to mutations in these genes mutations, and how the alteration of dynamic gene expression responds to the H1N1 virus induction is largely unidentified.
Early detection of H1N1 promotes individual treatment decisions and provides early data for predicting epidemics. However, early diagnosis of influenza is difficult in clinical practice (Memoli et al., 2008). The currently available for diagnosis of influenza infections, including viral isolation in cell culture, immunofluorescence assays, nucleic acid amplification tests, immunochromatography-based rapid diagnostic tests. But they are less practical in resource-limited regions due to their high cost and highly trained professionals (Vemula et al., 2016). Moreover, most of these influenza tests can detect and distinguish influenza A virus from influenza B virus, but the ability to further subtype influenza A virus is limited (Malanoski and Lin, 2013; Vemula et al., 2016). Furthermore, the recent newer diagnostic approaches, such as gene expression signatures in the peripheral blood and molecular surveillance of putative virulence factors in the experimental stage and were characterized by uncertainty (Azar and Landry, 2018). Therefore, accurate and early diagnosis of influenza viral infections are critical for the enhancement of HIN1 clinical management. Here, we used different samples to determine the gene expression difference between 21 h before infection and 77 h post-infection to construct an infectious diagnostic model using the multi-dimensional bioinformatics analysis. Subsequently, a diagnostic model was constructed, which showed good diagnostic performance for infections associated with the H1N1 virus. The genes from this model were then associated with the influenza virus infection, biological function, and infection-related reversal drugs. The study flowchart is illustrated in Fig. S1. This study could provide new insight for correlating genetic biomarkers for the research on the H1N1 virus pathogenesis and its clinical application.
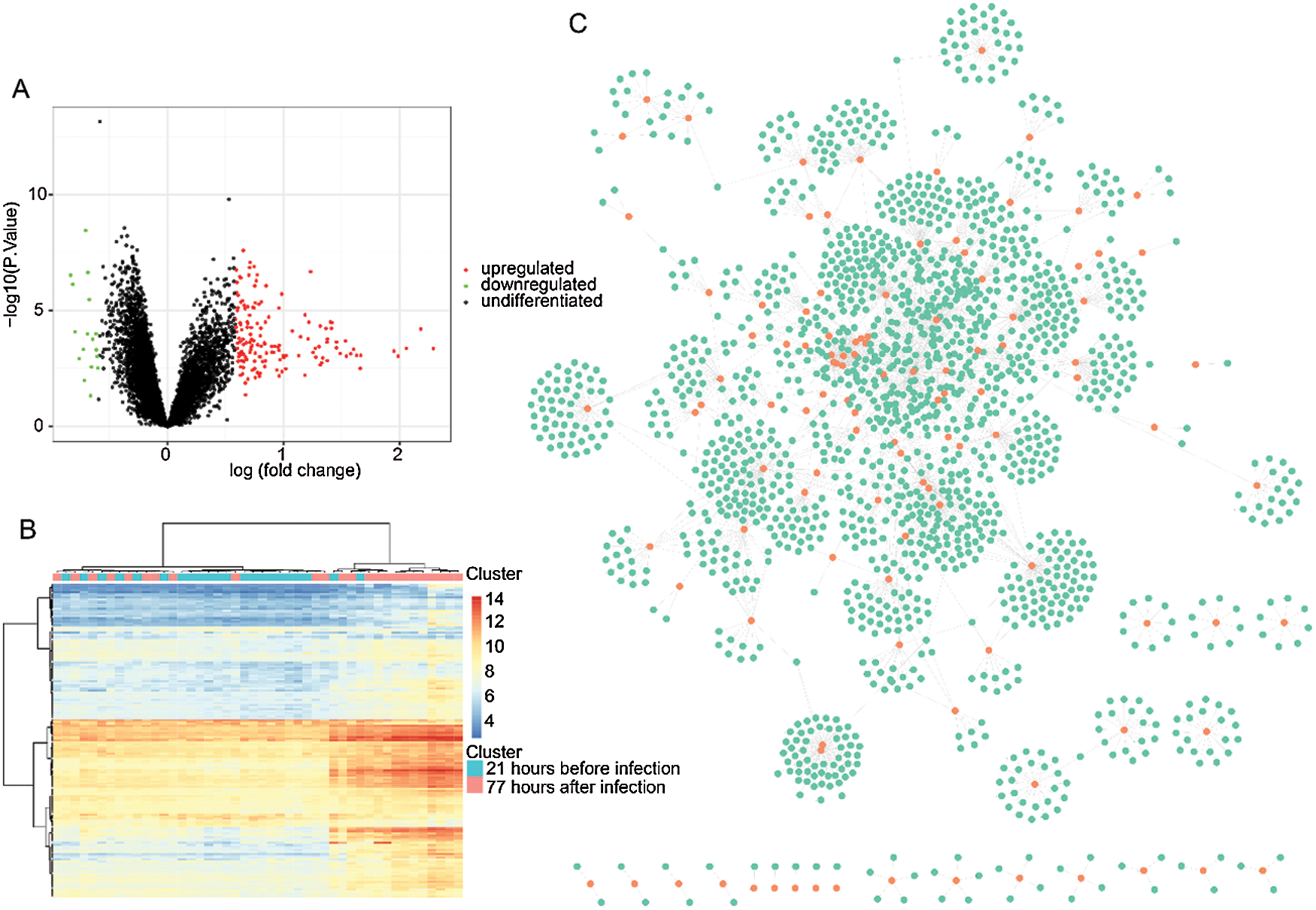
Figure 1: Identification of differentially expressed genes and PPI analysis.
Data set information and genes analysis
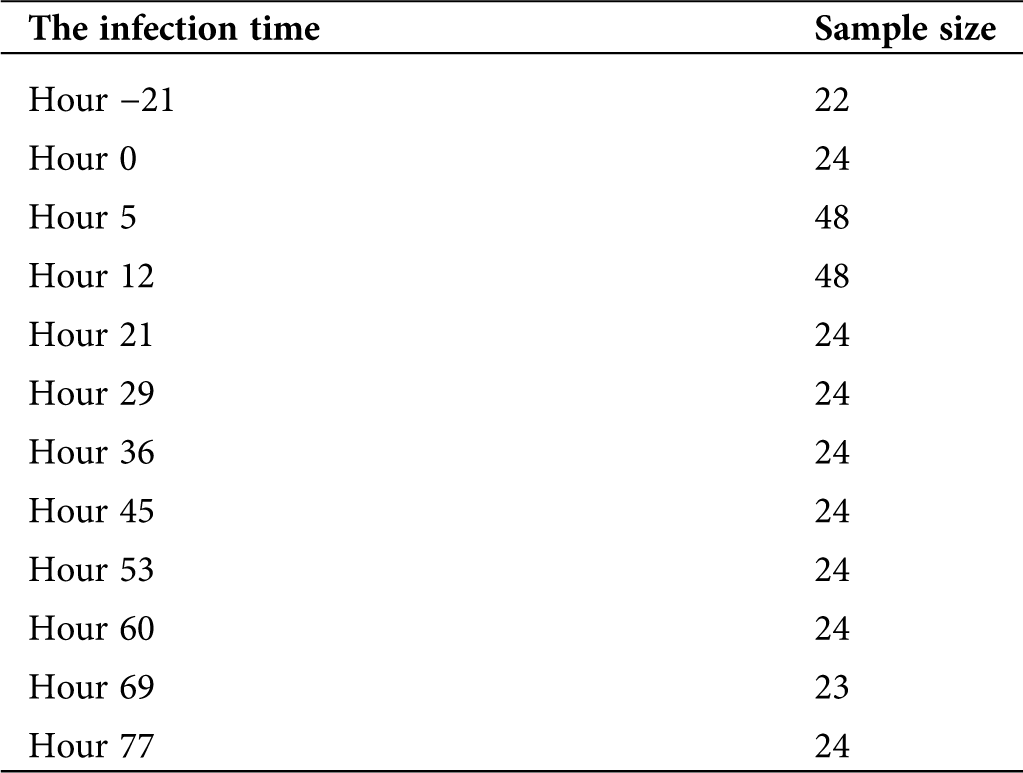
The data set comparing gene expression profiles before and after the manifestation of the H1N1 virus infection were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) (GSE73072). The dataset (GSE73072) from the GEO database was designed by Duke University under a contract awarded by the DARPA Predicting Health and Disease program. During each challenge study, subjects had peripheral blood taken prior to inoculation with the virus (baseline), immediately prior to inoculation (pre-challenge), and at set intervals following challenge. RNA was extracted at Expression Analysis from whole blood. The information details of GSE73072 could be referred to the website: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73072. The samples of infectious time used in this research were represented in Tab. 1. All statistical analyses on the gene expression data were analyzed using the Linear Models for Microarray (limma) package in R language, and only genes with a fold change > 1.5 or < 0.67 and P-value < 0.05 were considered to be differentially expressed. Subsequently, the differently expressed gene sets were imported into the STRING database (https://string-db.org/) and run using the protein-protein interaction (PPI) network. Next, the PPI network was constructed using the Cytoscape software. The STRING database (http://string-db.org) aims to provide a critical assessment and integration of protein-protein interactions, including direct (physical) as well as indirect (functional) associations. The detailed procedure of the use of the STRING database could be referred to in Damian et al. manuscript (Szklarczyk et al., 2015).
Table 1: Analysis of the clinical information of the samples
WGCNA analysis for the selection of infectious modules
The WGCNA R package was used to construct the weighted gene co-expression network of the differently expressed genes. WGCNA is a systems biology analysis method to construct scale-free networks using gene expression data. First, the similarity matrix was constructed using the gene expression data, which means calculating the absolute value of the Pearson correlation coefficient between two genes with the following formula:
Functional analysis of key modules
The molecular function of key modules was determined based on Gene Ontology (GO) analyses using the clusterProfiler R package and P < 0.05 taken as the threshold for statistical significance between groups. The associational network of the biological function items and genes was depicted using the Cytoscape software.
Identification of diagnostic model
Here, we applied a random forest algorithm to construct a diagnostic model based on the main genes used to predict the H1N1 virus infection. This random forest algorithm was executed, using the randomForest R package with a threshold of ntree = 500. Random forest is an ensemble classification algorithm integrating multiple decision trees through the idea of integrated learning. Its basic unit is the decision tree, whereby each tree depends on a random vector, and all these vectors in the random forest are independent and identically distributed. The area under the curve (AUC) was used to define the diagnostic prediction. Lastly, the diagnostic model gene expression levels between 1-, 2-, and 3-days post-infection were calculated using the analysis of Variance (ANOVA).
The diagnostic model genes related to the screening of candidate molecule drugs
To discover novel candidate therapeutic drugs for the anti-H1N1 virus, we selected individual up-regulated genes from the diagnostic model and mapped them to the connectivity map (CMAP) to investigate the cellular function of genes. The CMAP shows an effective matching algorithm, and the connected graph may significantly enrich the true positive molecule drug indication gene (Cheng et al., 2014). To identify the similarity, a calculation of the expression profiles of both the submitted genes and each drug-treated cell line was done. Here, the higher the similarity, the higher the consistency between the gene alternation and the submitted genes after the drug treatment.
H1N1 virus infection-related gene selection and WGCNA analysis
The estimated mean human incubation period for most influenza A virus infections is 1–3 days long, and within 3–4 days of post-infection, these viruses elicit strong inflammatory responses, which causes the clinical manifestations (Li and Cao, 2017). Therefore, to reveal HINI infection-associated genetic changes in humans, we analyzed the differentially expressed gene profiles between 22 samples of 21 h exposure time course before infection and 24 samples of 77 h exposure time course after infection of samples. As illustrated in Fig. 1A, 169 differently expressed genes were identified, in which 152 of them were upregulated, and 17 were downregulated. The heat cluster map analysis showed that those genes represented a significant difference between the two cohorts (Fig. 1B). Consequently, the 169 genes were subjected to PPI analysis, and as shown in Fig. 1C, we obtained a network that contained a total of 1725 genes and 2561 gene-gene interaction pairs.
Then, the WGCNA analysis was used to examine the co-expression modules of the 1725 genes. This algorithm was performed based on an unsigned topological overlap matrix, which applied a soft threshold power of β = 3 to exhibit an approximate scale-free topology (Fig. 2A). Also, as illustrated in Fig. 2B, these 1725 genes were allocated to 7 different modules according to their degree of connectivity. According to the standard of the hybrid dynamic shear tree, the minimum number of genes required in each gene network module was set at 20. Subsequently, we calculated the Pearson correlation coefficient (PC) and the P-value of the corresponding correlation for each module to determine the correlation between the gene modules and the infection caused by the H1N1 virus. The result illustrated in Fig. 2C showed that different modules such as green (PC = 0.55, P = 7e−05), turquoise (PC = 0.54, P = 1e−04), and yellow (PC = 0.65, P = 1e−06) were significantly associated with the H1N1 virus infection. Lastly, we calculated the gene significance (GS) value for each module, and the results, as shown in Fig. 2D, showed that the GS of green, turquoise, and yellow were significantly higher than that of other modules.
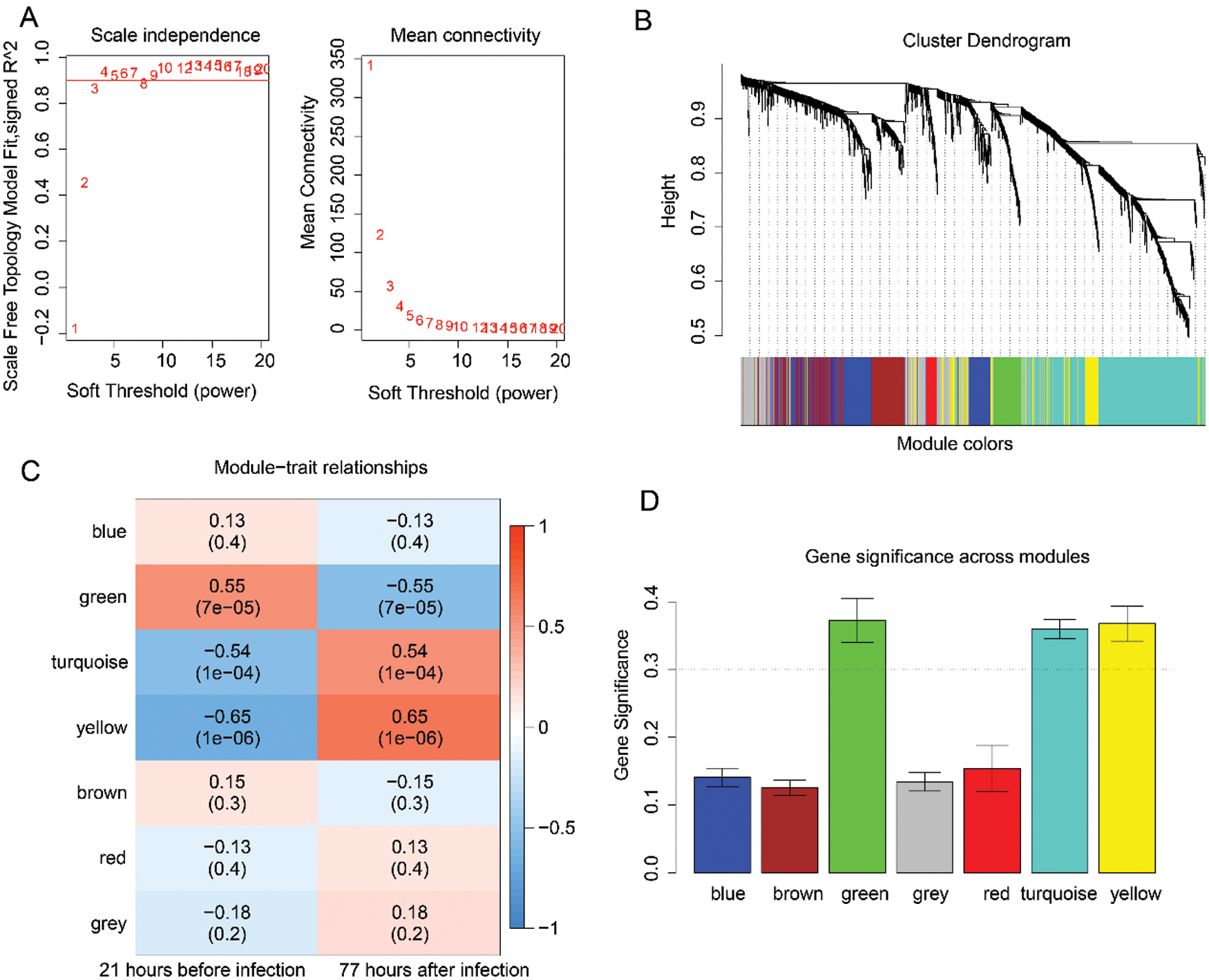
Figure 2: Analysis of the significant modules based on H1N1 virus infection-related genes using the WGCNA analysis.
Biological function of 3 modules
Here, we calculated the different degrees of nodes in each network module according to the gene expression relationships in the above 3 co-expression modules. It was observed that a small number of these genes had an obvious concentration, and their changes in expression will co-express with other adjacent nodes through interaction, which affects the downstream biological function. Therefore, these genes with higher node degrees are probably the key genes involved during HCC development. Hence, we only selected the top 20 genes with a higher node degree as the key gene in the co-expression network of each module. Subsequently, we carried out the GO analysis on all the 3 modules (including 60 genes) to explore the biological function of these genes. The biological function of 60 genes was depicted in Tab. S1. As illustrated in Fig. 3A, the genes of these 3 modules were significantly enhanced in virus infectious function, like the Type I interferon signaling, cellular response to Type I interferon, and response to Type I interferon. Lastly, Fig. 3B shows the interaction network of the biological pathways-genes which signified that the several biological functions were interacting with each other via some key genes such as OAS3, OAS1, IFNB1, and MX1.
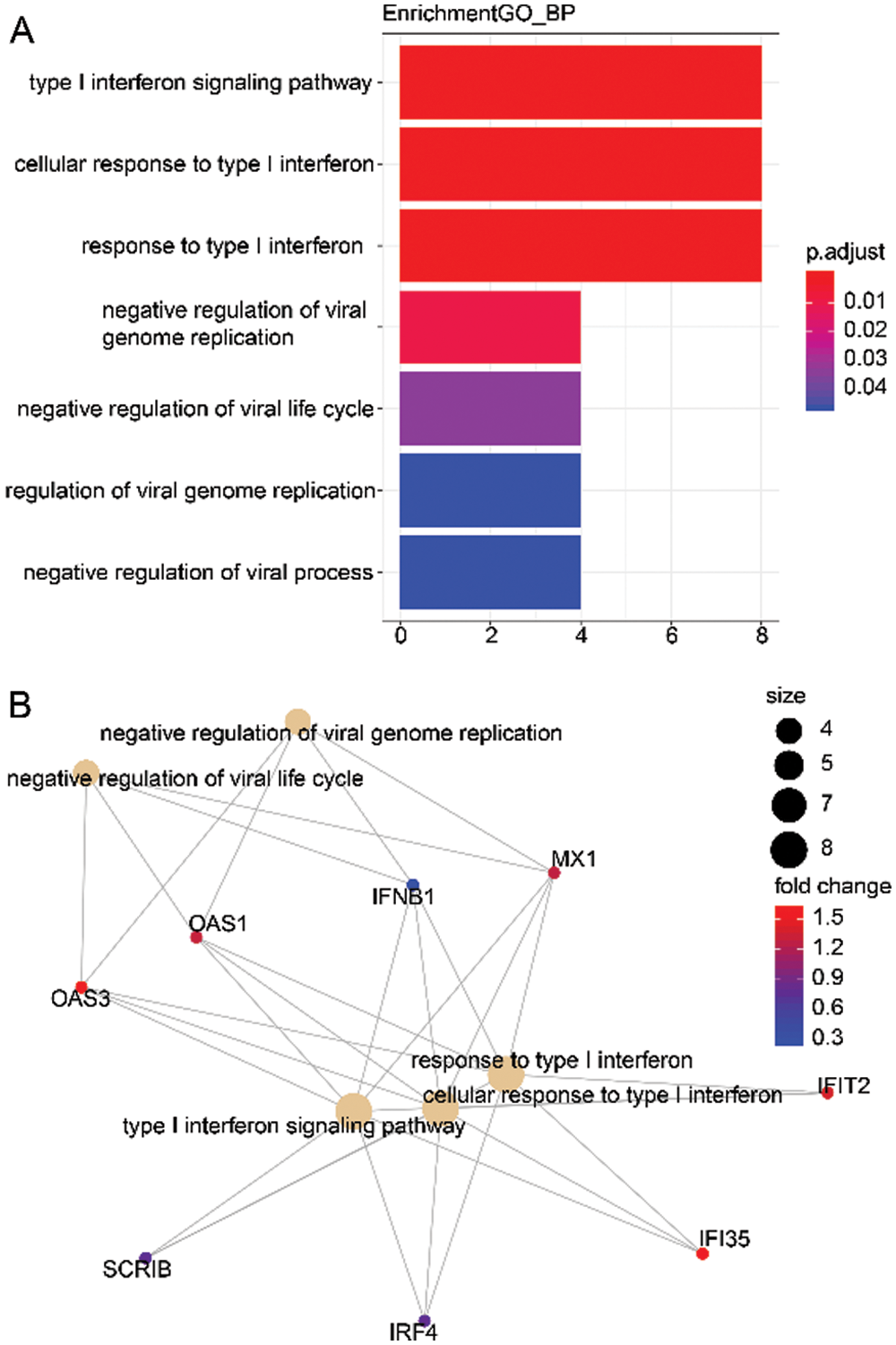
Figure 3: Biological function of 3 modules (60 genes).
Construction of prognostic model and validation
Based on the 60 core genes, we used a random forest algorithm test to construct the H1N1 infection diagnostic model. As shown in Fig. 4A, a comparison was done between before infection and 77 h post-infection time course to examine the differently expressed levels of the 60 genes in the infected samples, and particularly only the 20 genes were significantly represented in cluster heap. Then, the diagnostic model was used to verify 3 validation datasets, which included 22 samples of before infection and 120 samples of 1-day post-infection, 120 samples of 1-day post-infection, and 72 samples of 2-days post-infection, and 72 samples of 2-days post-infection and 47 samples of 3-days post-infection. The AUC of the receiver operating characteristic curve showed that the diagnostic model for 0–24 h, 24–48 h, and 48–72 h of infection, were 0.773, 0.794, and 0.831, respectively (Fig. 4B). Additionally, we analyzed the expression levels of the 20 genes across 1, 2, and 3 days. As illustrated in Fig. 5, it was noted that all the expression levels of these genes were upregulated with the increase in the progression of infection days (P < 0.05). These results proposed that the prognostic model made up of 60 genes were a potential biomarker for the diagnosis of infection caused by the H1N1 virus.
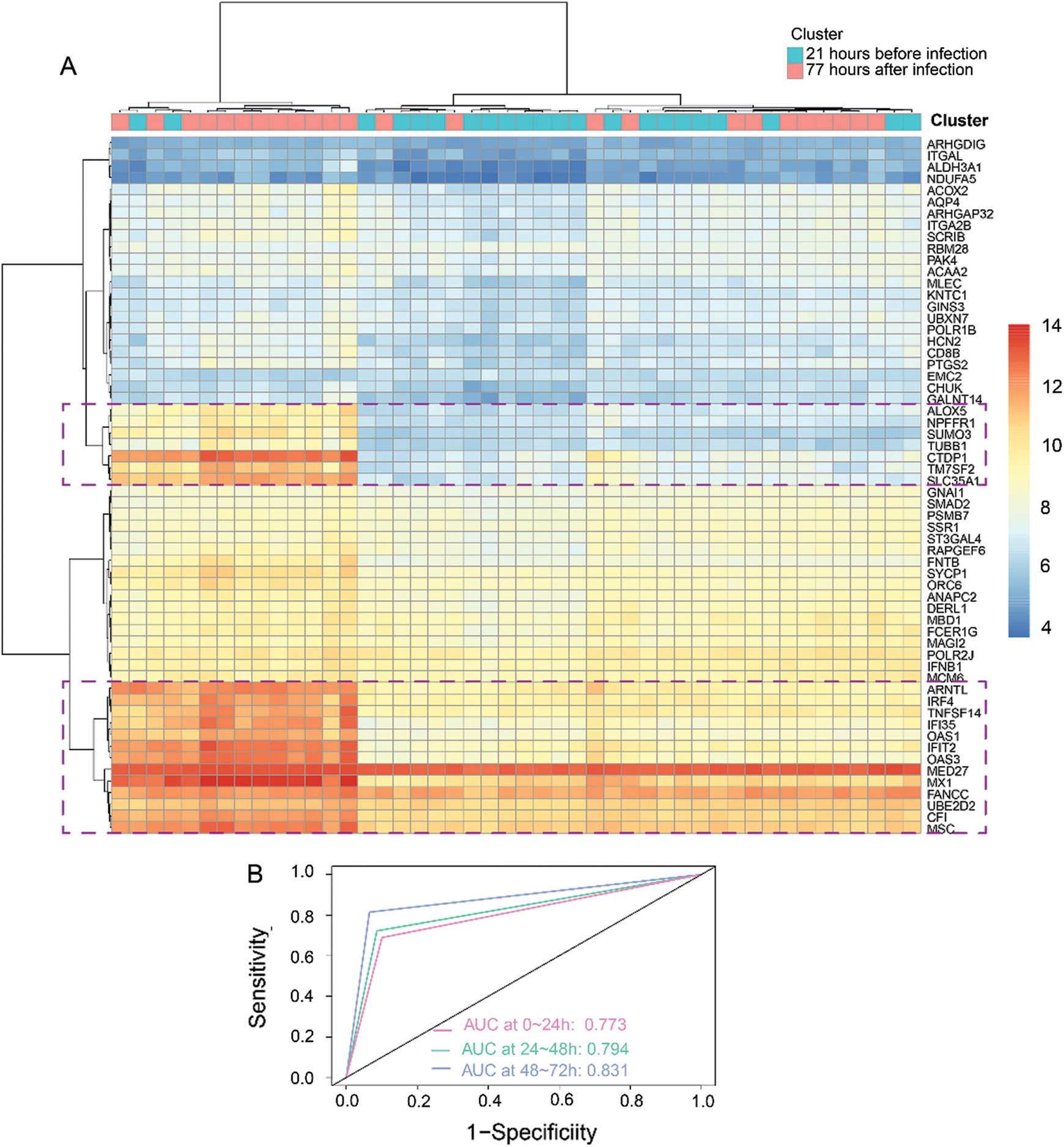
Figure 4: Construction and validation of a diagnostic model based on 3 modules (60 genes).
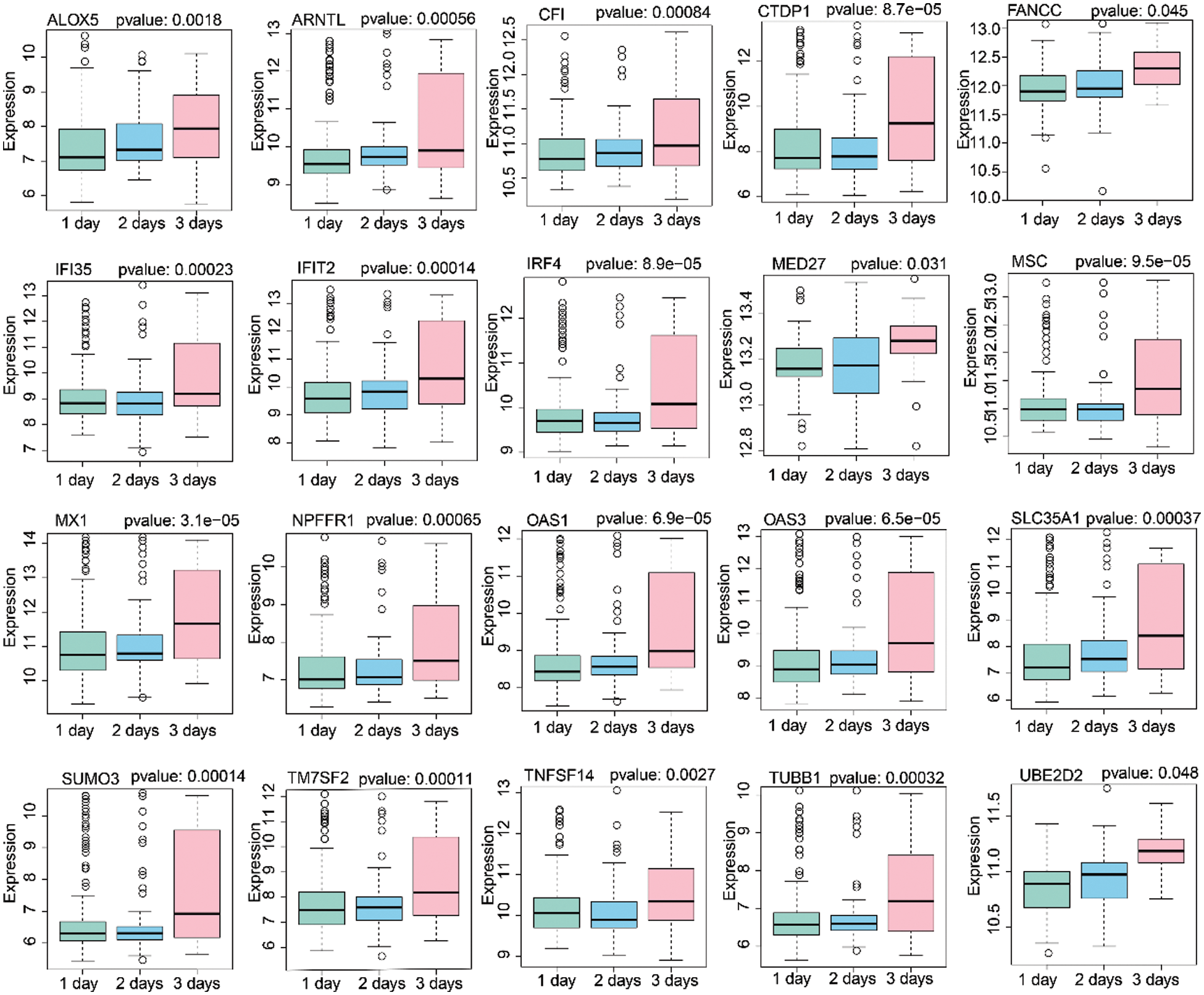
Figure 5: Analysis of 20 gene expression levels after 1 day, 2 days, and 3 days H1N1 virus infection.
Identification of gene-related drug molecular for anti-H1N1 virus
A total of 36 upregulated genes were selected from 60 different gene expressions to search for the infection-related reversal molecular drugs. This was achieved by calculating the different expression profiles of the submitted genes and each drug-treated cell line. Notably, many molecular drugs such as rimantadine, interferons, shikimic acid, and ribavirin, which are clinically used to treat influenza were observed to be associated with genes of the prognostic model (Tab. 2). These results indirectly validated that the prognostic model made up of 60 genes had a potential clinical application value in the treatment of infections caused by the H1N1 virus.
Table 2: Identification of gene-related drug molecular for anti-H1N1 virus

The time interval between the entry of the influenza pathogen into the human body and the onset of the earliest clinical manifestations is referred to as the incubation period (Clark and Lynch, 2011). Due to the very-short incubation period of the influenza virus, the time course before and after the onset of an illness is also very-short. Hence, influenza patients lack obvious clinical manifestations, and the progression of this disease can lead to its aggravation or become a potential source of infection (Rewar et al., 2015). Therefore, early identification of influenza latent infected patients is an effective measure that prevents its outbreak. Here, we constructed a diagnostic model for the H1N1 virus infection process based on the characteristic of the H1N1 virus infection-related genes using different multi-dimensional bioinformatics analysis procedures. We established that the diagnostic model was a potential biomarker for the diagnosis of the infection caused by the H1N1 virus. Our study examined the molecular biomarker for the H1N1 virus infection through bioinformatics methods, which provides a new view for studying the underlying mechanism of influenza infection and finding its clinical target.
The Influenza A virus is characterized based on genetic diversity and its susceptibility to different genetic mutations. However, much of our understanding of the influenza evolution comes from different sequencing analyses within a certain pathological range. This not only represents significant viral lineage changes within each infected host cell but also explains little information on the extent of genetic alternation after the host infection (Poon et al., 2016). To reveal this genetic alternation on the host after infection with the H1N1 virus, we made different expression analysis on the genes of samples obtained between 21 h before infection and 77 h post-infection. Subsequently, we selected the two-time nodes as the research object largely because the first day is the initial time of infection, whereas the third day is the most obvious period of generalized clinical manifestations after infection while avoiding the sample with severe clinical symptoms. Next, using the multi-dimensional bioinformatics analysis, we not only identified 60 genes from the 3 modules that proved to be potential biomarkers for the diagnosis of infection caused by the H1N1 virus but also we established that among these genes, 20 of them were associated with the progression of the HIN1 virus infectious. These findings are consistent with previous studies, which investigated that these 20 genes are involved in the pathogenesis of infections caused by the influenza virus. Horio et al. (2020) discovered that the ALOX5 gene was associated with the regulation of influenza virus replication through the daidzein activated signal transduction pathway, which indicated that it is an antiviral activity target. Elsewhere, the MX1 was shown to encode a guanosine triphosphate (GTP)-metabolizing protein that plays a role in the cellular antiviral response. Additionally, it could restrict the replication of the influenza A virus by targeting distinct sites of its nucleoprotein (Fatima et al., 2019). However, most of the gene mutations are not specific to H1N1, just like the annually genetic variation of influenza could not be fixed in a specific gene (Davidson, 2018). Moreover, much less is known about the genetic control of virus-host interactions in H1N1 disease (To et al., 2015). Herein, unlike these previous studies that established that a single gene was involved in the mechanism of viral infection, this diagnostic model that we constructed is composed of multiple genes, which can overcome various shortcomings such as lack of specificity and stability and susceptibility to external factors.
Besides, we used multidimensional bioinformatics to construct a H1N1 diagnostic model and analyze the biological function and drug targets of model genes. Bioinformatics has a wider application scope, a more complete coverage, and a more refined division of labor. The development of bioinformatics provides new ideas for the study of viruses and genes (Oulas et al., 2019). Moreover, our study complemented previous bioinformatics studies on H1N1. Liu et al. (2016) performed multi-dimensional algorithms to investigate a large scale of gene microarray data to constructed viral challenge model, which could predict each subject’s state of infection, including exposed nor infected, exposed but not infected, pre-acute phase of infection, acute phase of infection, post-acute phase of infection. Whereas our diagnostic model was aimed to found out patients with early H1N1 infection. A comprehensive analysis of our results and those of previous studies further contributes to mining out targeted genes associated with HIN1 infection and their clinical value.
In this study, the biological function of these 60 genes was also analyzed, and it was discovered that the Type I interferon and viral genome replication were the main gene enrichment items. In recent years, many research groups have adopted many different strategies to search for host factors involved in the virus genome replication cycle (Fan et al., 2019). The study of viral proteins related to the replication process of the influenza virus genome and the continuous discovery of virus-host interaction have contributed us to understand more accurately the life cycle of the influenza virus and the development of anti-influenza drugs (Jorba et al., 2009). Such as RNA polymerases, which are required for the genome transcription and replication of influenza A virus, have become ideal targets for the development of anti-influenza drugs (Du et al., 2018). Besides, gene fragment exchange and mismatch during replication in the virus genome was the main pathogenesis involved during mutation in viruses (Lowen, 2017). However, due to the complexity of genetic interactions between viral gene fragments, our understanding of the mechanisms involved during virus genome replication, which leads to mutations, remains inadequate (White and Lowen, 2018). Therefore, these results may provide new genetic targets for identifying these viral replication mutations, particularly during molecular interactions with host genes after viral infection. Moreover, our gene-related drug molecular association analysis found out that several clinical universal drug molecules for the therapy of influenza indirectly proved that this diagnostic model was an effective biomarker for the diagnosis of the H1N1 virus infection. For instance, we disclosed that the drug molecular analysis of shikimic acid inhibits the activity of neuraminidase and promotes the activity of important molecules that inhibit the activity of the influenza virus. Of note, the shikimic acid is a precursor of the synthetic drug ostamivir phosphate, which is an effective inhibitor used to treat a variety of seasonal influenza viruses (Martínez et al., 2015). Hence, the results of this study can provide genetic target support for the study and establishment of anti-influenza drugs.
Comparing to previous similar studies, our study has both shortcomings and advantages. Zaas et al. (2009) conducted a systematic statistical analysis to develop a blood mRNA expression signature that classifies symptomatic human respiratory viral infection. Whereas we use the multi-dimensional bioinformatics analysis method to build the diagnostic model, which is more advanced and comprehensive. In addition, our diagnostic models of genes were performed for biological analysis to further clarify the pathogenesis of these genes in H1N1. However, in contrast to Zaas et al. (2009) and Liu et al. (2016) studies, the associated analysis between the diagnostic model and influenza infectious stage in our study was not enough. Liu et al. (2016) constructed viral challenge model, which could predict each subject’s state of infection, including exposed nor infected, exposed but not infected, pre-acute phase of infection, acute phase of infection, post-acute phase of infection. And Zaas et al. (2009) not only included live rhinovirus, respiratory syncytial virus, and influenza A in their study, but also developed the gene signatures to classify human respiratory viral infection, such as symptomatic, asymptomatic, infected, and uninfected samples. One of the advantages of our study was a large scale of HIN1 infection samples were included in the investigation, which could overcome experimental errors and deviations. Whiles, our results still need to be validated in actual influenza samples. The study carried by Liu et al. (2016) and Zaas et al. (2009) verified their gene signature in peripheral blood for serum and plasma from infectious samples. Given this, the clinical application of the diagnostic model in our study is not very clear and needs further validation. We suggested that combining our research with previous research results may be more helpful to study the clinical targets of influenza infection.
In conclusion, this study has constructed a diagnostic model based on the H1N1 virus infection-related genes and has verified that this model is a potential biomarker for the diagnosis of the infection caused by the H1N1 virus. Besides, our research provides an integrated procedure for the discovery of gene characteristics and their corresponding biological function during the infection caused by the H1N1 virus, and to also establish drug targets against anti-influenza.
Author Contribution: The authors confirm contribution to the paper as follows: Study conception and design: Hongyan Diao; data collection: Wenbiao Chen, Kefan Bi, Jingjing Jiang, Xujun Zhang; analysis and interpretation of results: Wenbiao Chen, Kefan Bi, Jingjing Jiang; draft manuscript preparation: Hongyan Diao, Wenbiao Chen. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: All data was collected from the public database.
Funding Statement: This work was supported by the major national S&T projects for infectious diseases (2018ZX10301401), the Key Research & Development Plan of Zhejiang Province (2019C04005), the National Key Research, and the Development Program of China (2018YFC2000500).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Azar MM, Landry ML (2018). Detection of influenza A and B viruses and respiratory syncytial virus by use of Clinical Laboratory Improvement Amendments of 1988 (CLIA)-waived point-of-care assays: A paradigm shift to molecular tests. Journal of Clinical Microbiology 56: e00367-18. DOI 10.1128/JCM.00367-18. [Google Scholar] [CrossRef]
Clark N, Lynch J (2011). Influenza: Epidemiology, clinical features, therapy, and prevention. Seminars in Respiratory and Critical Care Medicine 32: 373–392. DOI 10.1055/s-0031-1283278. [Google Scholar] [CrossRef]
Cheng J, Yang L, Kumar V, Agarwal P (2014). Systematic evaluation of connectivity map for disease indications. Genome Medicine 6: 540. DOI 10.1186/s13073-014-0095-1. [Google Scholar] [CrossRef]
Cheng Z, Zhou J, To KK, Chu H, Li C, Wang D, Yang D, Zheng S, Hao K, Bossé Y, Obeidat M, Brandsma CA, Song YQ, Chen Y, Zheng BJ, Li L, Yuen KY (2015). Identification of TMPRSS2 as a susceptibility gene for severe 2009 pandemic A(H1N1) influenza and A(H7N9) influenza. Journal of Infectious Diseases 212: 1214–1221. DOI 10.1093/infdis/jiv246. [Google Scholar] [CrossRef]
Davidson S (2018). Treating influenza infection, from now and into the future. Frontiers in Immunology 9: 517. DOI 10.3389/fimmu.2018.01946. [Google Scholar] [CrossRef]
Du Y, Xin L, Shi Y, Zhang TH, Wu NC, Dai L, Gong D, Brar G, Shu S, Luo J, Reiley W, Tseng YW, Bai H, Wu TT, Wang J, Shu Y, Sun R (2018). Genome-wide identification of interferon-sensitive mutations enables influenza vaccine design. Science 359: 290–296. DOI 10.1126/science.aan8806. [Google Scholar] [CrossRef]
Fan H, Walker AP, Carrique L, Keown JR, Serna Martin I, Karia D, Sharps J, Hengrung N, Pardon E, Steyaert J, Grimes JM, Fodor E (2019). Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 573: 287–290. DOI 10.1038/s41586-019-1530-7. [Google Scholar] [CrossRef]
Fatima U, Zhang Z, Zhang H, Wang XF, Xu L, Chu X, Ji S, Wang X (2019). Equine Mx1 restricts Influenza A virus replication by targeting at distinct site of its nucleoprotein. Viruses 11: 1114. DOI 10.3390/v11121114. [Google Scholar] [CrossRef]
Fineberg HV (2014). Pandemic preparedness and response-lessons from the H1N1 influenza of 2009. New England Journal of Medicine 370: 1335–1342. DOI 10.1056/NEJMra1208802. [Google Scholar] [CrossRef]
Horio Y, Sogabe R, Shichiri M, Ishida N, Morimoto R, Ohshima A, Isegawa Y (2020). Induction of a 5-lipoxygenase product by daidzein is involved in the regulation of influenza virus replication. Journal of Clinical Biochemistry and Nutrition 66: 36–42. DOI 10.3164/jcbn.19-70. [Google Scholar] [CrossRef]
Jorba N, Coloma R, Ortín J (2009). Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathogens 5: e1000462. DOI 10.1371/journal.ppat.1000462. [Google Scholar] [CrossRef]
Josset L, Belser JA, Pantin-Jackwood MJ, Chang JH, Chang ST, Belisle SE, Tumpey TM, Katze MG (2012). Implication of inflammatory macrophages, nuclear receptors, and interferon regulatory factors in increased virulence of pandemic 2009 H1N1 influenza A virus after host adaptation. Journal of Virology 86: 7192–7206. DOI 10.1128/JVI.00563-12. [Google Scholar] [CrossRef]
Langfelder P, Horvath S (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. DOI 10.1186/1471-2105-9-559. [Google Scholar] [CrossRef]
Li H, Cao B (2017). Pandemic and avian Influenza A viruses in humans: Epidemiology, virology, clinical characteristics, and treatment strategy. Clinics in Chest Medicine 38: 59–70. DOI 10.1016/j.ccm.2016.11.005. [Google Scholar] [CrossRef]
Liu TY, Burke T, Park LP, Woods CW, Zaas AK, Ginsburg GS, Hero AO (2016). An individualized predictor of health and disease using paired reference and target samples. BMC Bioinformatics 17: 47. DOI 10.1186/s12859-016-0889-9. [Google Scholar] [CrossRef]
Lowen AC (2017). Constraints, drivers, and implications of Influenza A virus reassortment. Annual Review of Virology 4: 105–121. DOI 10.1146/annurev-virology-101416-041726. [Google Scholar] [CrossRef]
Lutz Iv MM, Dunagan MM, Kurebayashi Y, Takimoto T (2020). Key role of the Influenza A virus PA gene segment in the emergence of pandemic viruses. Viruses 12: 365. DOI 10.3390/v12040365. [Google Scholar] [CrossRef]
Lyons DM, Lauring AS (2018). Mutation and epistasis in Influenza virus evolution. Viruses 10: 407. DOI 10.3390/v10080407. [Google Scholar] [CrossRef]
Malanoski AP, Lin B (2013). Evolving gene targets and technology in influenza detection. Molecular Diagnosis & Therapy 17: 273–286. DOI 10.1007/s40291-013-0040-9. [Google Scholar] [CrossRef]
Martínez JA, Bolívar F, Escalante A (2015). Shikimic acid production in Escherichia coli: From classical metabolic engineering strategies to omics applied to improve its production. Frontiers in Bioengineering and Biotechnology 3: 145. [Google Scholar]
Memoli MJ, Morens DM, Taubenberger JK (2008). Pandemic and seasonal influenza: Therapeutic challenges. Drug Discovery Today 13: 590–595. DOI 10.1016/j.drudis.2008.03.024. [Google Scholar] [CrossRef]
Oulas A, Minadakis G, Zachariou M, Sokratous K, Bourdakou MM, Spyrou GM (2019). Systems bioinformatics: Increasing precision of computational diagnostics and therapeutics through network-based approaches. Briefings in Bioinformatics 20: 806–824. DOI 10.1093/bib/bbx151. [Google Scholar] [CrossRef]
Poon LL, Song T, Rosenfeld R, Lin X, Rogers MB, Zhou B, Sebra R, Halpin RA, Guan Y, Twaddle A, DePasse JV, Stockwell TB, Wentworth DE, Holmes EC, Greenbaum B, Peiris JSM, Cowling BJ, Ghedin E (2016). Quantifying influenza virus diversity and transmission in humans. Nature Genetics 48: 195–200. DOI 10.1038/ng.3479. [Google Scholar] [CrossRef]
Rewar S, Mirdha D, Rewar P (2015). Treatment and prevention of pandemic H1N1 influenza. Annals of Global Health 81: 645–653. DOI 10.1016/j.aogh.2015.08.014. [Google Scholar] [CrossRef]
Sun Y, Wang Q, Yang G, Lin C, Zhang Y, Yang P (2016). Weight and prognosis for influenza A(H1N1)pdm09 infection during the pandemic period between 2009 and 2011: A systematic review of observational studies with meta-analysis. Infectious Diseases 48: 813–822. DOI 10.1080/23744235.2016.1201721. [Google Scholar] [CrossRef]
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C (2015). STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Research 43: D447–452. DOI 10.1093/nar/gku1003. [Google Scholar] [CrossRef]
To KKW, Zhou J, Chan JF, Yuen KY (2015). Host genes and influenza pathogenesis in humans: An emerging paradigm. Current Opinion in Virology 14: 7–15. DOI 10.1016/j.coviro.2015.04.010. [Google Scholar] [CrossRef]
Uyeki TM, Bernstein HH, Bradley JS, Englund JA, File TM, Fry AM, Gravenstein S, Hayden FG, Harper SA, Hirshon JM, Ison MG, Johnston BL, Knight SL, McGeer A, Riley LE, Wolfe CR, Alexander PE, Pavia AT (2019). Clinical practice guidelines by the Infectious Diseases Society of America: 2018 update on diagnosis, treatment, chemoprophylaxis, and institutional outbreak management of seasonal influenzaa. Clinical Infectious Diseases 68: e1–e47. DOI 10.1093/cid/ciy866. [Google Scholar] [CrossRef]
Vemula SV, Zhao J, Liu J, Wang X, Biswas S, Hewlett I (2016). Current approaches for diagnosis of influenza virus infections in humans. Viruses 8: 96. DOI 10.3390/v8040096. [Google Scholar] [CrossRef]
White MC, Lowen AC (2018). Implications of segment mismatch for influenza A virus evolution. Journal of General Virology 99: 3–16. DOI 10.1099/jgv.0.000989. [Google Scholar] [CrossRef]
Zaas AK, Chen M, Varkey J, Veldman T, Hero III AO, Lucas J, Huang Y, Turner R, Gilbert A, Lambkin-Williams R, Øien NC, Nicholson B, Kingsmore S, Carin L, Woods CW, Ginsburg GS (2009). Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host & Microbe 6: 207–217. DOI 10.1016/j.chom.2009.07.006. [Google Scholar] [CrossRef]
Zeldovich KB, Liu P, Renzette N, Foll M, Pham ST, Venev SV, Gallagher GR, Bolon DN, Kurt-Jones EA, Jensen JD, Caffrey DR, Schiffer CA, Kowalik TF, Wang JP, Finberg RW (2015). Positive selection drives preferred segment combinations during influenza virus reassortment. Molecular Biology and Evolution 32: 1519–1532. DOI 10.1093/molbev/msv044. [Google Scholar] [CrossRef]
Supplementary Figure
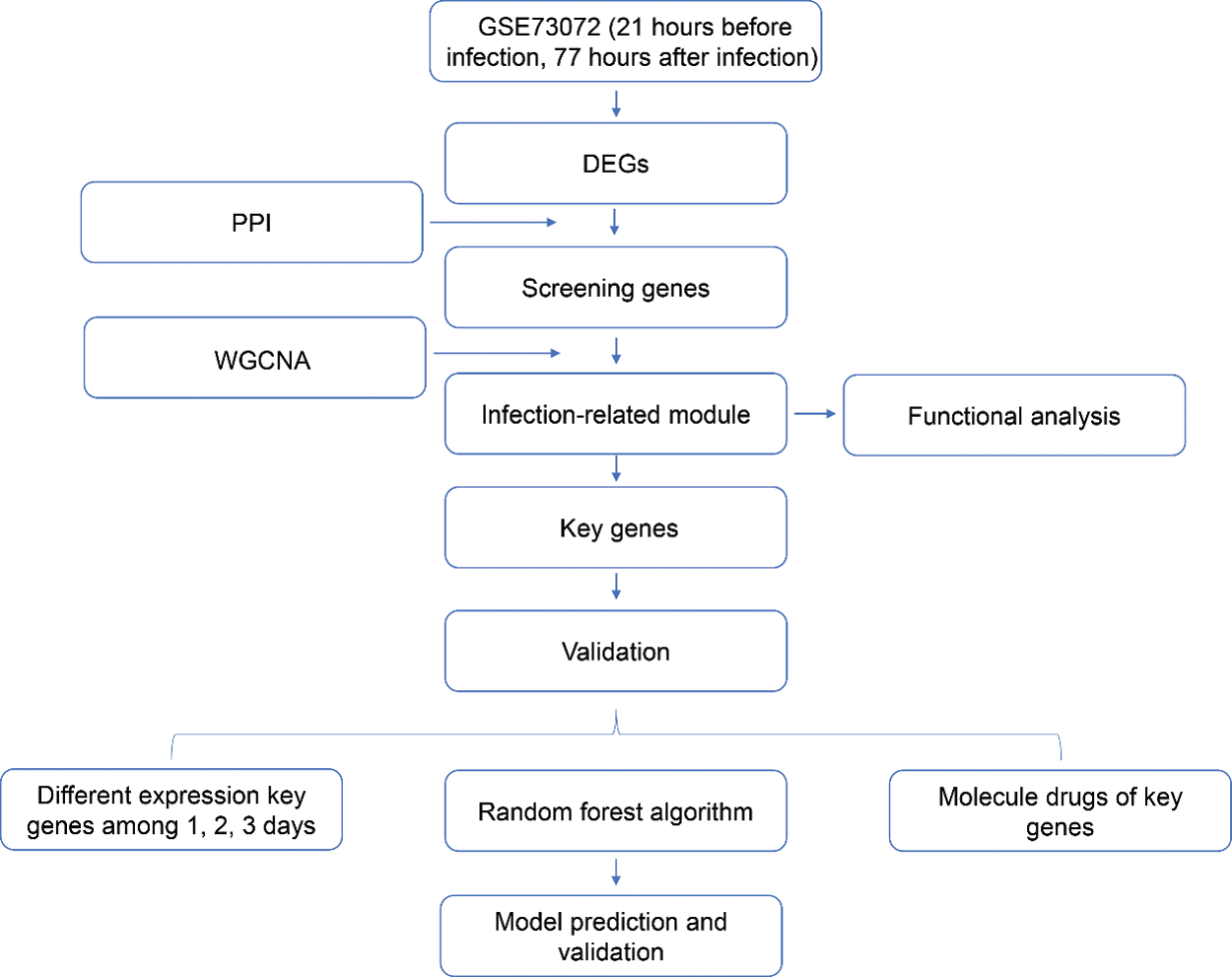
Figure S1: The flow chart of this research.
Supplementary Table
Table S1: Biological function of 60 genes

 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |