DOI:10.32604/biocell.2021.015530
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.015530 | www.techscience.com/journal/biocell |
| Article |
PPARγ LBD and its ligand specificity reveal a selection of potential partial agonist: Molecular dynamics based T2D drug discovery initiative
1Department of Applied Science, Galgotias College of Engineering and Technology, Knowledge Park-II, Greater Noida, 201306, India
2Institute for Skeletal Aging & Orthopedic Surgery, Hallym University-Chuncheon Sacred Heart Hospital, Chuncheon-si, 24252, Korea
3Department of Zoology, Fakir Mohan University, Vyasa Vihar, Balasore, 756020, India
4Department of Biotechnology, School of Life Science and Biotechnology, Adamas University, Kolkata, 700126, India
*Address correspondence to: Sang-Soo Lee, 123sslee@gmail.com; Chiranjib Chakraborty, drchiranjib@yahoo.com
Received: 25 December 2020; Accepted: 03 February 2021
#These authors contributed equally to this work
Abstract: PPARγ is a peroxisome proliferator-activated receptor (PPAR) family protein and is a target for type 2 diabetes (T2D). In this paper, we have performed a molecular docking analysis between ligand molecules (CID9816265, CID11608015, CID20251380, CID20251343, CID20556263, CID624491, CID42609928, and CID86287562) and PPARγ to determine the ligand specificity. It also helps to understand the ligand-binding domain (LBD) activity of PPARγ during the binding of the ligand. Further, a molecular dynamics simulation study was performed to determine the ligand biding stability in the PPARγ LBD. Its ligand specificity informed us about the potentiality of selecting a partial agonist. The study also shows the binding conformation of Ceramicine B having hydrogen bonding affinity with a tricyclic polar head and stabilized the β-sheet region. On the other hand, the tricyclic polar head of nimbolide also formed hydrogen bonding (Ser342), but it shows a lesser degree of stabilization in the β-sheet region. It shows the binding conformation of partial agonist (PPARγ) in the Pocket-II of PPARγ LBD, which has a significant role in stabilizing the β-sheet region. It might help to regulate ERK/Cdk5 mediated phosphorylation of Ser245. The study helps us understand the valid pose of a set of ligands confirmation and target protein conformation using docking and molecular dynamics study. This in silico study will also help to initiate a drug discovery process of T2D.
Keywords: PPARγ; Partial agonist; Molecular simulation; T2D; Drug discovery
PPAR (peroxisome proliferator-activated receptors) family proteins belong to nuclear receptor superfamily proteins. The proteins of the superfamily control an extensive range of lipid-associated-genes accountable for lipid metabolism, cholesterol metabolism, lipid transport, and adipose differentiation. These proteins regulate energy homeostasis (Bain et al., 2007; Cox, 2017). PPARs are mainly found in the nucleus. However, PPARs are transported in between cytoplasm and nucleus whenever is needed (Umemoto and Fujiki, 2012). Three diverse family members from PPAR superfamilies are PPARα, PPARβ, and PPARγ. PPARβ is also known as PPARδ. These diverse family members/subtypes are highly homologous (Kroker and Bruning, 2015). It has been noted that PPARα is expressed in the tissues and is highly metabolically active, especially in the kidney, heart, liver, and muscle. PPARα is mainly associated with lipid metabolism (Kersten and Stienstra, 2017; Zoete et al., 2007). PPARβ or PPARδ is related to different diseases like obesity, diabetes, heart disease, and metabolic hypertension (Feige et al., 2006; Jiang et al., 2019; Yang and Long, 2018). Conversely, PPARγ is found to be expressed in the adipose tissues. It has been found that PPARγ can help in the creation of minute insulin-sensitive adipocytes.
Therefore, it has a function as a regulator in insulin sensitization. So, PPARγ is considered as a target for treating T2D (Type 2 diabetes) (Leonardini et al., 2009; Jiang et al., 2020).
PPARγ consists of 5 domains, which are named as A to E and are shown in Fig. 1a. These domains are distributed in the direction from N- to C-terminus. These domains are highly conserved and comparable to other nuclear receptor proteins. (i) A and B domain: Both of these domain contains AF1 (activation function 1). It engrosses with the ligand-free coregulator binding (Diezko and Suske, 2013). (ii) C domain: This domain contains DNA binding domain and is highly conserved. It is responsible for DNA binding (IJpenberg et al., 1997). (iii) Hing domain: This domain helps in the rotation between the domain C and E (the DNA binding domain (C) and the ligand-binding domain (E)). It is a poorly conserved domain. (iv) Ligand binding domain (LBD)/activation function 2 (AF2): It contains a ligand-binding pocket and helps in the ligand binding. This domain is highly conserved (Kroker and Bruning, 2015). Endogenous and synthetic ligands can bind to the ligand-binding pocket (LBP), which is situated within the ligand-binding domain (LBD). The LBP has three ligand-binding branches with different properties and ligand specificity, resulting in a Y-shaped cavity for the ligand to bind. Three branches (I, II & III) are depicted in Fig. 1b. Branch-I is hydrophilic in nature, and molecules with an acidic head easily bind in this pocket, formed by H3, H5, H11 & H12 helices. Ligands (full agonists) binding in Branch-I strongly stabilizes H12 of LBD, which leads to transcriptional activation of PPAR (Gampe, 2000) by allowing the co-activators to dock. H3, Ω-loop, and β-sheet region form Branch-II; this pocket is of hydrophobic character. Ligand binding to this region shows weak interaction with the residues from Branch-I, rather stabilizing the β-sheet region and showing transcriptional activity (Malapaka et al., 2012; Zheng et al., 2013; Delfosse et al., 2015; Amato et al., 2012). Branch-III consists of both hydrophobic and hydrophilic regions, formed by β-sheet, H2, H3, and H5 helices.

Figure 1: (a) Different domain of PPARγ protein. (b) Ligand binding domain (LBD) of PPARγ; Different branches of ligand binding pocket are as mentioned in Roman letter. The 13 α-helices of LBD are also labelled.
It has been noted that full agonists of PPARγ can attach with the Branch-I of the LBP, and, is responsible for the formation of hydrogen bonds with the different residues such as Ser289, His323, His449, and Tyr473 (Nolte et al., 1998). Thiazolidinediones (TZDs) are an example of full agonists that were used to treat T2D. However, unwanted side effects have been noted during the use of full agonists. The side effects include fluid retention, fat accumulation, loss of bone density, cancer, augmented risk of heart failure, etc. (Kung and Henry, 2012; Nissen and Wolski, 2007; Rubenstrunk et al., 2007; Tang and Maroo, 2007). Due to the side effects, full agonists are not used for the treatment of T2D. On the other hand, recent studies reported several chemical compounds that bind in the Branch-II of PPAR LBP between H3, Ω-loop, and β-sheet. These compounds are partial agonists (Amato et al., 2012; Choi et al., 2010; Kroker and Bruning, 2015), and does not show full PPARγ agonism but block the ERK/Cdk5 (cyclin-dependent kinase 5) mediated PPARγ phosphorylation at Ser245 (Ser273 for PPARγ2) (Banks et al., 2015). Blocking of PPARγ phosphorylation leads to dysregulated expression of a series of genes, including adiponectin and adipsin. It regulates insulin sensitivity with diminished adverse effects (Dunn et al., 2011; Higgins and DePaoli, 2010; Yi et al., 2017). For these reasons, instead of full agonists of PPAR, nowadays partial agonists of PPARγ are the choice for new generation anti-diabetic drugs. Our recent finding also reports Ceramicine with particular moiety could bind Branch-II of PPARγ LBD (Mallick, 2018).
Here we report the virtual screening of small molecules from PubChem Database to find the structure of the molecules that will occupy the ligand-binding pocket between H3 and β-sheet (Branch-II). Naphthalene group of a partial agonist SR2067 has been reported to stabilize the β-sheet region of Branch-II employing hydrophobic interaction between helix 3 and the β-sheet (Maltsev and Oswald, 2010). In the present study, we focused on searching for the bicyclic/tricyclic/tetracyclic molecules to find their binding affinity in LBP of PPARγ. These bicyclic/tricyclic/tetracyclic molecules used for the present study are ((CID9816265, CID11608015); (CID20251380, CID20251343); (CID20556263, CID624491); CID42609928 (Ceramicine B), CID86287562 (Nimbolide)). Further, large molecules with the bicyclic/tricyclic head and different hydrophobic tails have also been searched from the PubChem database to ensure if these molecules can show specific binding modes to act as PPARγ partial agonists. The present study reveals the ligand specificity of ligand binding pocket of PPARγ during binding of the ligand. Its ligand specificity informed us about the potentiality of the selection of partial agonists. Additionally, a molecular interactions study has been executed to determine the ligand-binding affinity with PPARγ LBD as well as binding stability. This in silico study also initiates the drug discovery of T2D.
PPARγ structure and ligand compounds
We have used the X-ray crystal structure of PPARγ, which is an isoform of this receptor protein. The structure was collected from RCBS Protein Data Bank (PDB) and the PDB ID 2F4B (Mahindroo et al., 2006). The unwanted atoms and bound ligand were removed from the PPARγ protein structure and minimized using the GROMACS-4.5 software package (Pronk et al., 2013). The structures of ligand compounds (Fig. 2) were collected from the PubChem compound database, and the structure was optimized with the help of Chimera software.

Figure 2: The structure of the ligand compounds used for our study (Bicyclic molecules (CID9816265, CID11608015); Tricyclic molecules (CID20251380, CID20251343); Tetra cyclic molecules (CID20556263, CID624491); CID42609928 (Ceramicine B), CID86287562 [Nimbolide]) [The ligand structure was adopted from PubChem].
Molecular docking study and virtual screening study
The docking simulation studies of the different ligand molecules in PPARγ LBD were performed with AutoDock4 (Morris et al., 2009). AutoDock is extensively used docking software. This software uses a Lamarckian genetic algorithm to analyze docking between protein and ligand (Hou et al., 2013). The protein was kept as rigid molecules and the ligand as flexible molecules for generating a protein-ligand complex using AutoDock software. A cubic grid box was created of size 60 Å × 60 Å × 60 Å to encompass all the active site residues of PPARγ. At the finishing step of each docking process, the lowest energy conformation was considered the best binding conformation between each considered molecule and PPARγ.
Software LIGPLOT was used to understand the hydrophobic and hydrogen bond contacts between the protein and ligand (Laskowski and Swindells, 2011; Wallace et al., 1995).
Molecular dynamic (MD) simulation
We have performed MD simulations using the GROMACS-4.5.6 package (CDAC server, India) (Hess et al., 2008). All-atom molecular dynamics simulations of PPARγ protein with each ligand inhibitors were performed using this software package under the GROMOS force field. The force field parameter was laid down as 53A6 (Mallick et al., 2019; Oostenbrink et al., 2004; van Gunsteren et al., 1996).
The PPARγ protein with each ligand inhibitor complex was handled alone by putting them into a cubic box. All the complexes were solvated by using the SPC216 water model. To electrically neutralize the total charge, Na ions were added to each system by using the genion tool of the GROMACS package. The system replaces water molecules with ions at the most constructive electrostatic potential positions at random. Finally, we performed energy minimization using the steepest descent algorithm with a maximum step size of 0.01 nm. During energy minimization, we have maintained a tolerance of 1000 kJ/mol/nm. The applied system was restrained equilibration, and the parameter was placed as LINCS (linear constraint solver for molecular simulations) constraints for all bonds and at 300 K and 1 bar for 100 ns for heavy atoms (Hess et al., 1997).
Molecular docking study and virtual screening study
We have performed molecular docking of PPARγ with the different ligand compounds (Bicyclic molecules (CID9816265, CID11608015); Tricyclic molecules (CID20251380, CID20251343); Tetra cyclic molecules (CID20556263, CID624491); and large molecule with tricyclic head CID42609928 (Ceramicine B), CID86287562 (Nimbolide). The structure of the docking complexes for the selected molecules in PPARγ LBD is depicted in Fig. 3. Fig. 3a shows ligand-binding pocket in the presence of bicyclic molecules, which are CID9816265 and CID11608015. We have found main binding residues are PHE226, PRO227, LEU228, ARG288, GLU291, GLU295, and GLU343. The molecules CID9816265 and CID11608015 have binding energies of −7.65 kcal/mol and −7.46 kcal/mol for their lowest energy conformation. Fig. 3b shows ligand-binding pocket in the presence of tricyclic molecules, which are CID20251380 and CID20251343. This ligand-binding pocket includes some active binding residues, which are GLU291, ARG288, LEU228, LEU333, GLU343, LEU340, SER342, and ILE341. The lowest binding energy for tricyclic molecules CID20251380 and CID20251343 in PPARγ LBD are −8.38 kcal/mol and −7.92 kcal/mol, respectively. Fig. 3c depicts the ligand-binding pockets in the presence of tetracyclic molecules, which are CID20556263 and CID624491. This ligand-binding pocket includes some active binding residues, which are GLU291, ARG288, LEU333, PHE287, THR268, GLU343, SER342, and ILE341. The best binding conformation for tetracyclic molecules CID20556263 and CID624491 has binding energy −8.2 kcal/mol and −8.63 kcal/mol.
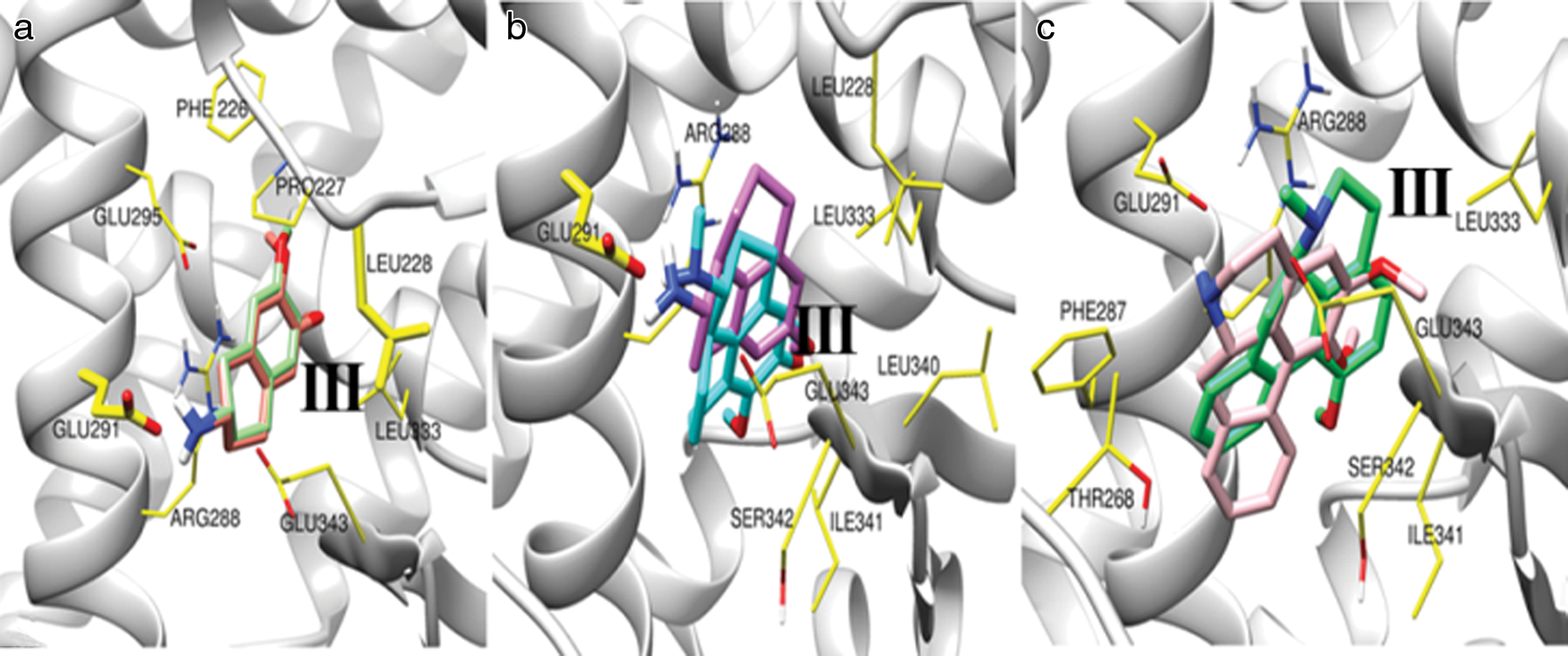
Figure 3: The local structure of the docking complexes for selected molecules in PPARγ LBD.
The docking complexes structure between CID42609928 (Ceramicine B) and PPARγ is shown in Fig. 4. Fig. 4a portrays the binding pose of the large molecule Ceramicine B in PPARγ LBD. The binding energy for the lowest energy conformation was found to be −9.2 kcal/mol. We have noted that molecule CID42609928 has a tricyclic polar head. Fig. 4b displays hydrophobic and hydrogen bond contacts between PPARγ protein and CID42609928 (Ceramicine B) compound determined by the LIGPLOT program. This ligand-binding pocket includes some active binding residues, which are ARG288, CYS285, PHE287, GLE284, MET384, SER342, GLU343, LEU270, THR268, and ILE281. The LIGPLOT program also shows identical active ligand-binding residues.

Figure 4: The structure of the docking complexes for CID42609928 (Ceramicine B) in PPARγ LBD.
The structure of the docking complexes of CID86287562 (Nimbolide) and PPARγ is shown in Fig. 5. Fig. 5a informs us about the local structure of the docking complex for the large molecule nimbolide with PPARγ. The lowest energy for binding confirmation is −9.42 kcal/mol. We have noted that molecule CID86287562 has a tricyclic polar head. Fig. 5b shows hydrophobic and hydrogen bond contacts between PPARγ protein and CID86287562 (Nimbolide) compound as determined by the LIGPLOT program. This ligand-binding pocket includes some active binding residues, which are ARG288, LEU330, LEU333, GLU343, SER342, MET364, ILE341, MET348, and CYS385. The LIGPLOT program also informed the same active ligand-binding residues.

Figure 5: The structure of the docking complexes for CID86287562 (Nimbolide) in PPARγ LBD.
Root-mean-square deviation of the PPARγ with the different ligands
Binding conformations of CID86287562 (Nimbolide) and CID42609928 (Ceramicine B) in LBD of PPARγ is similar to the other partial agonist reported in the previous studies (Amato et al., 2012; Motani et al., 2009). Thus molecular dynamics simulation study has been performed for PPARγ-CID86287562 (Nimbolide) and PPARγ-CID42609928 (Ceramicine B) complexes to determine their interaction under dynamic conditions. RMSD value of the Cα atoms fluctuations of PPARγ protein for these complexes is compared with uncomplexed PPARγ protein in Fig. 6 for 100 ns simulation. The simulation of three molecules shows more or less the same pattern of fluctuations with lower RMSD value for Cα atoms fluctuations of PPARγ in complex with CID42609928 (Ceramicine B) molecule. The Cα-RMSD for all three cases shows a steady increase up to 10 ns and then stabilized after 40 ns, during the simulation.

Figure 6: The RMSD value of the Cα atoms during 100 ns simulation of PPARγ (red), PPARγ -CID86287562/Nimbolide (green) and PPARγ -CID42609928/Ceramicine B (black) system.
Root-mean-square fluctuation of the PPARγ with the different ligand
Fig. 7 represents RMSF of Cα atoms of PPARγ as a function of amino acids for the three considered complexes PPARγ, PPARγ-CID86287562 (Nimbolide), and PPARγ-CID42609928 (Ceramicine B) during 100 ns simulation. The RMSF value over the entire simulation was plotted against residue numbers and has been displayed in this figure. The strength of fluctuation of the PPARγ-CID42609928 (Ceramicine B) complex has reduced the RMSF value for H3 and β-sheet relative to other considered complexes. Also, displacement of H12 for PPARγ-CID42609928 (Ceramicine B) complex is comparable to uncomplexed one, whereas highly stabilized RMSF is observed for PPARγ-CID86287562 (Nimbolide) complex. Thus, RMSF variation indicates a binding mode of CID42609928 (Ceramicine B), stabilizing H3, β-sheet, and Ser245 region more strongly than the binding mode of PPARγ-CID86287562 (Nimbolide) complex. Stabilization of these regions helps in blocking the ERK/Cdk5-mediated phosphorylation of Ser245. Further, dynamics of H12 of PPARγ completed with CID86287562 (Nimbolide) seems affected, whereas it remains unaffected in the binding orientation of CID42609928 (Ceramicine B) molecule, which could reflect H12 independent transcriptional activity of PPARγ by CID42609928 molecule (Gelman et al., 2007; Lu et al., 2006).

Figure 7: The RMSF of Cα atoms as a function of amino acids for PPARγ(red), PPARγ– complexed with CID86287562/Nimbolide (green) and PPARγ– complexed with CID42609928/Ceramicine B (black) system.
Here, we have applied molecular docking studies to find the small molecule that will occupy the ligand-binding pocket between H3 and β-sheet of PPARγ. A specific configuration of these particular molecules do the job of sandwich filling between Arg288, Glu291 of H3 and Ser342, Glu343 of β-sheet; consequently, the polar atom of the ligands easily forms hydrogen-bonding networks with the residues of the β-sheet, which preferentially will stabilize the β-sheet region of the LBD of PPARγ protein. Further, large molecules with similar specific configurations also show high binding affinity in the Pocket II of LBD, but the orientation of the molecule changes with the configuration of the hydrophobic tail. Thus, a Molecular dynamics simulation study was performed to establish the orientation of hydrophobic tail of large molecule in Pocket II that will inhibit Ser273 phosphorylation with a lesser degree of H12 stabilization.
Molecular docking was desired to forecast the optimal ligand-receptor complex orientation and conformation using two steps. The first step act through the assembly conformation of ligands in the active site of the protein. The second step helps the positioning of the conformations through a scoring function to forecast binding rigidity for each orientation (Meng et al., 2011).
The LIGPLOT helps to understand the interaction regions and simultaneous interaction points. It also illustrated the interaction of residues through hydrogen bonds and hydrophobic contacts. Finally, the interactions (H bond) are pointed out by a dashed (–) line between the two atoms involved in this interaction. It was shown in our previous study also (Chakraborty et al., 2014). Similarly, hydrophobic interactions are shown through an arc with spokes radiating toward the ligand atoms.
Virtual screening of small molecule uses MTiOpenScreen server, which uses chemical libraries from PubChem Database. It reveals that the bicyclic/tricyclic region of the molecules align between helix 3 and the β-sheet of PPARγ LBD (results are not shown here). However, the β-sheet plays an essential role in PPARγ LBD. A recent study suggested that the naphthalene group of a partial agonist SR2067 stabilized the β-sheet region through hydrophobic regions between helix 3 and the β-sheet (Maltsev and Oswald, 2010). Thus, virtual screening in-home PC was performed for the bicyclic/tricyclic molecules with and without polar residues to find their binding affinity in LBD of PPARγ. Also, large molecules with the bicyclic/tricyclic head have been searched from the PubChem database to ensure if these kinds of molecules can show specific binding modes to act as a PPARγ partial agonist. A docking simulation study can predict one molecule’s preferred orientation in the active site of a target protein. Therefore, we performed docking simulation to identify the binding affinity and orientation of each considered compound in PPARγ protein using AutoDock. Bicyclic molecules shown in Fig. 2 with PubChem CID9816265, CID11608015 have been docked in the ligand-binding pocket. Minimum energy docking conformation reveals that the methoxy group of both the molecules CID9816265 and CID11608015 form hydrogen bond with the residue LEU288 and amine group with GLU291. Though both the molecules show hydrophobic interaction with GLU343 of the β-sheet region, the molecules’ minimum energy binding conformation is not precisely a sandwich filling conformation between H3 and β-sheet. The minimum energy binding position of these molecules in the LBD is represented in Fig. 3a. The bicyclic molecule CID931 (Naphthalene), lacking polar atom, has the lowest energy binding conformation in parallel to H5 in ligand binding Pocket III. The binding conformation of this molecule is shown in Supplementary Fig. 1a.

Supplementary Figure 1: Lowest energy binding conformation of (a) bicyclic (CID931/Naphthalene) and (b) Tricyclic (CID 44358650) molecule in the ligand binding pocket of PPARγ. The binding energy of lowest energy conformation for CID931 & CID 44358650 are −5.52 kcal/mol and −6.77 kcal/mol respectively.
Increasing a cyclic ring in the bicyclic molecule changes the orientation to its lowest energy docking conformation in the binding pocket. The binding conformation of the tricyclic molecule CID20251380 and CID20251343 in PPARγ LBD is depicted in Fig. 3b. Results show that these molecules’ minimum energy conformation moves towards the β-sheet to align between H3 and β-sheet. The amine group of these tricyclic molecules also form hydrogen bonding with GLU291 and more residues from the β-sheet region involved in hydrophobic interaction. Increasing another cyclic ring also allows these types of molecules to have minimum energy binding conformation in Pocket II of the LBD (Fig. 3c). In order to check the binding affinity of tetracyclic molecules, compounds with CID20556263 and 624491 were taken from the PubChem database. A bicyclic head characterizes the above-considered molecules with polar residues at the opposite side of the ring (Positions 1, 2, 5, or 6). The side chains of the polar residues GLU291, THR268, GLU343, and α-amino group of SER342 allow these kinds of molecules to have the lowest binding energy in Pocket-II. The nitrogen atom of these molecules aligns towards the side chain of GLU291, while the oxygen atom of the bicyclic head towards the SER342. Thus, the molecules’ binding pattern with polar bicyclic head has a sandwich filling pose between H3 and β-sheet in the large “Y” shaped ligand-binding cavity of PPARγ protein, and polar oxygen could form hydrogen bond interaction with key residue SER342. It is important to mention here, similar molecule with bicyclic head without the polar atom at both the end have lowest energy binding conformation parallel to H5 in ligand binding pocket. The binding conformation of these molecules in the LBP is shown in Supplementary Fig. 1b.
As these fragment-like compounds have a lower binding affinity and can move from the binding site in dynamic conditions, docking simulation studies were performed for large molecules with tricyclic polar head. Large molecule CID86287562 (Nimbolide) and CID42609928 (Ceramicine B) have been considered for our study. These molecules have a tricyclic head with a polar atom at the opposite end and a large hydrophobic tail. The lowest energy binding mode reveals that the molecules fit in Pocket II in parallel with H3, polar head forms a hydrogen bond with the α-amino group of SER342, as depicted in Fig. 4. Their binding confirmation reveals that all the molecules fit in the Pocket II parallel with H3, polar head forms a hydrogen bond with the α-amino group of SER342. Moreover, the tail region’s remaining hydrophobic interaction spreads out to the bottom (binding pose 1) or top (binding pose 2) of Pocket II depending on the molecule’s configuration.
Studies have shown that the β-sheet region has a significant role in stabilizing the LBD of PPARγ, which results in the blocking of ERK/CDK5 mediated phosphorylation. Therefore, β-sheet plays a vital role in acting as a partial agonist of PPARγ. The β-sheet region is stabilized by means of hydrogen bonding, where an acidic group has a vital role, and most PPARγ partial agonists act through the β-sheet region to stabilize the complex structure. There is a significant role of acidic groups, which has been shown in β-sheet stabilization. It also has a hydrogen-binding network to the different backbone atoms of the β-sheet, especially the backbone nitrogen of Ser342 (van Marrewijk et al., 2016). Our study shows that the binding conformation of Ceramicine B has hydrogen bonding with the tricyclic polar head and stabilized the β-sheet region. On the other hand, the tricyclic polar head of Nimbolide also formed hydrogen bonding with Ser342, but it shows a lesser degree of stabilization of the β-sheet region. It shows that the binding conformation of partial agonist (PPARγ) in the Pocket II of PPARγ LBD has a significant role in stabilizing the β-sheet region and blocking ERK/Cdk5 mediated phosphorylation of Ser245 (van Marrewijk et al., 2016).
This study aims to foresee a valid pose from a receptor configuration and a set of ligand conformation through their scoring-based system depending on their binding affinity. The study could be used as scaffolds to discover more potent compounds or as positive controls for new screens. This study also helps to initiate the drug discovery of T2D. Based on the binding patterns and poses, several future works can be done to improve the biological activity of ligand binding pocket (LBP) inhibitors by increasing the number of the typical hydrogen bonds, molecular volume, and electrostatic interactions. We propose our in silico outcomes could be a valuable resource for pharmacologists as well as molecular biologists who are working in the drug development process. Considering these probable structural outcomes, linked to the PPARγ ligand-binding domain and its ligand specificity, the study will help future researchers to understand the selection of potential partial agonists.
Acknowledgement: This study was supported by Hallym University Research Fund and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1C1C1008694 & NRF-2020R1I1A3074575).
Availability of Data and Materials: All data and materials included within the manuscript.
Author Contribution: BM: Conceptualization, Data curation, Methodology, Software, Investigation. ARS: Visualization, Investigation, Writing-Review & Editing. MB: Visualization, Investigation, Writing-Review & Editing. S-SL: Writing-Review & Editing, Supervision. CC: Conceptualization, Writing-Original draft, Review & Editing, Supervision.
Ethics Approval: Not Applicable.
Funding Statement: The author(s) received no specific funding for this study.
Conflicts of Interest: The authors declare that they have no conflict of interest.
Amato Aélica A, Rajagopalan S, Lin JZ, Carvalho BM, Figueira ACM, Lu J, Ayers SD, Mottin M, Silveira RL, Souza PCT, Mourão RH V, Saad Mário J A, Togashi M, Simeoni LA, Abdalla Déia S P, Skaf MS, Polikparpov I, Lima MCA, Galdino SL, Brennan RG, Baxter JD, Pitta IR, Webb P, Phillips KJ, Neves FAR (2012). GQ-16, a novel peroxisome proliferator-activated receptor γ (PPARγ) ligand, promotes insulin sensitization without weight gain. Journal of Biological Chemistry 287: 28169–28179. DOI 10.1074/jbc.M111.332106. [Google Scholar] [CrossRef]
Bain DL, Heneghan AF, Connaghan-Jones KD, Miura MT (2007). Nuclear receptor structure: Implications for function. Annual Review of Physiology 69: 201–220. DOI 10.1146/annurev.physiol.69.031905.160308. [Google Scholar] [CrossRef]
Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, Spiegelman BM (2015). An ERK/Cdk5 axis controls the diabetogenic actions of PPARγ. Nature 517: 391–395. DOI 10.1038/nature13887. [Google Scholar] [CrossRef]
Chakraborty C, Hsu MJ, Agoramoorthy G (2014). Understanding the molecular dynamics of Type-2 diabetes drug target DPP-4 and its interaction with sitagliptin and inhibitor diprotin-A. Cell Biochemistry and Biophysics 70: 907–922. DOI 10.1007/s12013-014-9998-0. [Google Scholar] [CrossRef]
Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, Griffin PR (2010). Obesity-linked phosphorylation of PPARγ by cdk5 is a direct target of the anti-diabetic PPARγ ligands. Nature 466: 451–456. DOI 10.1038/nature09291. [Google Scholar] [CrossRef]
Cox RL (2017). Rationally designed PPARδ-specific agonists and their therapeutic potential for metabolic syndrome. Proceedings of the National Academy of Sciences of the United States of America 114: 3284–3285. DOI 10.1073/pnas.1702084114. [Google Scholar] [CrossRef]
Delfosse V, Le Maire A, Balaguer P, Bourguet WA (2015). A structural perspective on nuclear receptors as targets of environmental compounds. Acta Pharmacologica Sinica 36: 88–101. DOI 10.1038/aps.2014.133. [Google Scholar] [CrossRef]
Diezko R, Suske G (2013). Ligand binding reduces SUMOylation of the peroxisome proliferator-activated receptor γ (PPARγ) activation function 1 (AF1) domain. PLoS One 8: e66947. DOI 10.1371/journal.pone.0066947. [Google Scholar] [CrossRef]
Dunn FL, Higgins LS, Fredrickson J, DePaoli AM, Group IS (2011). Selective modulation of PPARγ activity can lower plasma glucose without typical thiazolidinedione side-effects in patients with Type 2 diabetes. Journal of Diabetes and its Complications 25: 151–158. [Google Scholar]
Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W (2006). From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Progress in Lipid Research 45: 120–159. DOI 10.1016/j.plipres.2005.12.002. [Google Scholar] [CrossRef]
Gampe RT,Jr., Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, Kliewer SA, Willson TM, Xu HE (2000). Asymmetry in the PPARγ/RXRα crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Molecular Cell 5: 545–555. DOI 10.1016/S1097-2765(00)80448-7. [Google Scholar] [CrossRef]
Gelman L, Feige JN, Desvergne B (2007). Molecular basis of selective PPARγ modulation for the treatment of Type 2 diabetes. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1771: 1094–1107. DOI 10.1016/j.bbalip.2007.03.004. [Google Scholar] [CrossRef]
Hess B, Bekker H, Berendsen HJ, Fraaije JG (1997). LINCS: a linear constraint solver for molecular simulations. Journal of Computational Chemistry 18: 1463–1472. [Google Scholar]
Hess B, Kutzner C, van der Spoel D, Lindahl E (2008). GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation 4: 435–447. DOI 10.1021/ct700301q. [Google Scholar] [CrossRef]
Higgins LS, dePaoli AM (2010). Selective peroxisome proliferator-activated receptor γ (PPARγ) modulation as a strategy for safer therapeutic PPARγ activation. American Journal of Clinical Nutrition 91: 267S–272S. DOI 10.3945/ajcn.2009.28449E. [Google Scholar] [CrossRef]
Hou X, Du J, Zhang J, Du L, Fang H, Li M (2012). How to improve docking accuracy of AutoDock4. 2: A case study using different electrostatic potentials. Journal of Chemical Information and Modeling 53: 188–200. DOI 10.1021/ci300417y. [Google Scholar] [CrossRef]
IJpenberg A, Jeannin E, Wahli W, Desvergne B (1997). Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA A functional analysis of the malic enzyme gene PPAR response element. Journal of Biological Chemistry 272: 20108–20117. DOI 10.1074/jbc.272.32.20108. [Google Scholar] [CrossRef]
Jiang Y, Li Q, Jia M, Yan Z (2019). PPARδ: a potential therapeutic target for the treatment of metabolic hypertension. International Journal of Hypertension 2019: 7809216. [Google Scholar]
Jiang H, Zhou XE, Shi J, Zhou Z, Zhao G, Zhang X, Sun Y, Suino-Powell K, Ma L, Gao H, Yu X (2020). Identification and structural insight of an effective PPARγ modulator with improved therapeutic index for anti-diabetic drug discovery. Chemical Science 11: 2260–2268. DOI 10.1039/C9SC05487A. [Google Scholar] [CrossRef]
Kersten S, Stienstra R (2017). The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 136: 75–84. DOI 10.1016/j.biochi.2016.12.019. [Google Scholar] [CrossRef]
Kroker AJ, Bruning JB (2015). Review of the structural and dynamic mechanisms of PPARγ partial agonism. PPAR Research 2015: 816856. [Google Scholar]
Kung J, Henry RR (2012). Thiazolidinedione safety. Expert Opinion on Drug Safety 11: 565–579. DOI 10.1517/14740338.2012.691963. [Google Scholar] [CrossRef]
Laskowski RA, Swindells MB (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling 51: 2778–2786. DOI 10.1021/ci200227u. [Google Scholar] [CrossRef]
Leonardini A, Laviola L, Perrini S, Natalicchio A, Giorgino F (2009). Cross-talk between PPAR and insulin signaling and modulation of insulin sensitivity. PPAR Research 2009: 818945. [Google Scholar]
Lu IL, Huang CF, Peng YH, Lin YT, Hsieh HP, Chen CT, Lien TW, Lee HJ, Mahindroo N, Prakash E, Yueh A, Chen HY, Goparaju CMV, Chen X, Liao CC, Chao YS, Hsu JTA, Wu SY (2006). Structure-based drug design of a novel family of PPARγ partial agonists: virtual screening, X-ray crystallography, and in vitro/in vivo biological activities. Journal of Medicinal Chemistry 49: 2703–2712. DOI 10.1021/jm051129s. [Google Scholar] [CrossRef]
Mahindroo N, Wang CC, Liao CC, Huang CF, Lu IL, Lien TW, Peng YH, Huang WJ, Lin YT, Hsu MC, Lin CH, Tsai CH, Hsu JTA, Chen X, Lyu PC, Chao YS, Wu SY, Hsieh HP (2006). Indol-1-yl acetic acids as peroxisome proliferator-activated receptor agonists: Design, synthesis, structural biology, and molecular docking studies. Journal of Medicinal Chemistry 49: 1212–1216. DOI 10.1021/jm0510373. [Google Scholar] [CrossRef]
Malapaka RR, Khoo S, Zhang J, Choi JH, Zhou XE, Xu Y, Gong Y, Li J, Yong EL, Chalmers MJ, Chang L (2012). Identification and mechanism of 10-carbon fatty acid as modulating ligand of peroxisome proliferator-activated receptors. Journal of Biological Chemistry 287: 183–195. DOI 10.1074/jbc.M111.294785. [Google Scholar] [CrossRef]
Mallick B (2018). Molecular dynamics simulations reveal the role of ceramicine B as novel PPARγ partial agonist against type 2 diabetes. arXiv preprint arXiv: 180808375. [Google Scholar]
Mallick B, Sharma AR, Lee SS, Chakraborty C (2019). Understanding the molecular interaction of human argonaute-2 and miR-20a complex: A molecular dynamics approach. Journal of Cellular Biochemistry 120: 19915–19924. DOI 10.1002/jcb.29300. [Google Scholar] [CrossRef]
Maltsev AS, Oswald RE (2010). Hydrophobic side chain dynamics of a glutamate receptor ligand binding domain. Journal of Biological Chemistry 285: 10154–10162. DOI 10.1074/jbc.M109.088641. [Google Scholar] [CrossRef]
Meng X, Zhang HX, Mezei M, Cui M (2011). Molecular docking: A powerful approach for structure-based drug discovery. Current Computer–Aided Drug Design 7: 146–157. DOI 10.2174/157340911795677602. [Google Scholar] [CrossRef]
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry 30: 2785–2791. DOI 10.1002/jcc.21256. [Google Scholar] [CrossRef]
Motani A, Wang Z, Weiszmann J, McGee LR, Lee G, Liu Q, Staunton J, Fang Z, Fuentes H, Lindstrom M, Liu J, Biermann DHT, Jaen J, Walker NPC, Learned RM, Chen JL, Li Y (2009). INT131: A selective modulator of PPARγ. Journal of Molecular Biology 386: 1301–1311. DOI 10.1016/j.jmb.2009.01.025. [Google Scholar] [CrossRef]
Nissen SE, Wolski K (2007). Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine 356: 2457–2471. DOI 10.1056/NEJMoa072761. [Google Scholar] [CrossRef]
Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV (1998). Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature 395: 137–143. DOI 10.1038/25931. [Google Scholar] [CrossRef]
Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF (2004). A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. Journal of Computational Chemistry 25: 1656–1676. DOI 10.1002/jcc.20090. [Google Scholar] [CrossRef]
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E (2013). GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29: 845–854. DOI 10.1093/bioinformatics/btt055. [Google Scholar] [CrossRef]
Rubenstrunk A, Hanf R, Hum DW, Fruchart JC, Staels B (2007). Safety issues and prospects for future generations of PPAR modulators. Biochimica et Biophysica Acta (BBA)–Molecular and Cell Biology of Lipids 1771: 1065–1081. [Google Scholar]
Tang WW, Maroo A (2007). PPARγ agonists: Safety issues in heart failure. Diabetes, Obesity and Metabolism 9: 447–454. DOI 10.1111/j.1463-1326.2006.00616.x. [Google Scholar] [CrossRef]
Umemoto T, Fujiki Y (2012). Ligand-dependent nucleo-cytoplasmic shuttling of peroxisome proliferator-activated receptors, PPARα and PPARγ. Genes to Cells 17: 576–596. DOI 10.1111/j.1365-2443.2012.01607.x. [Google Scholar] [CrossRef]
van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger PK (1996). Biomolecular Simulation: The GROMOS96 Manual and User Guide. Zürich: Vdf Hochschulverlag AG an der ETH Zürich 86: 1–1042. [Google Scholar]
van Marrewijk LM, Polyak SW, Hijnen M, Kuruvilla D, Chang MR, Shin Y, Kamenecka TM, Griffin PR, Bruning JB (2015). SR2067 reveals a unique kinetic and structural signature for PPARγ partial agonism. ACS Chemical Biology 11: 273–283. DOI 10.1021/acschembio.5b00580. [Google Scholar] [CrossRef]
Wallace AC, Laskowski RA, Thornton JM (1995). LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Engineering Design & Selection 8: 127–134. DOI 10.1093/protein/8.2.127. [Google Scholar] [CrossRef]
Yang Q, Long Q (2018). PPARδ, a potential therapeutic target for heart disease. Nuclear Receptor Research 5: 159. DOI 10.32527/2018/101375. [Google Scholar] [CrossRef]
Yi W, Shi J, Zhao G, Zhou XE, Suino-Powell K, Melcher K, Xu HE (2017). Identification of a novel selective PPARγ ligand with a unique binding mode and improved therapeutic profile in vitro. Scientific Reports 7: 645. DOI 10.1038/srep41487. [Google Scholar] [CrossRef]
Zheng W, Feng X, Qiu L, Pan Z, Wang R, Lin S, Hou D, Jin L, Li Y (2013). Identification of the antibiotic ionomycin as an unexpected peroxisome proliferator-activated receptor γ (PPARγ) ligand with a unique binding mode and effective glucose-lowering activity in a mouse model of diabetes. Diabetologia 56: 401–411. DOI 10.1007/s00125-012-2777-9. [Google Scholar] [CrossRef]
Zoete V, Grosdidier A, Michielin O (2007). Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1771: 915–925. DOI 10.1016/j.bbalip.2007.01.007. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |