DOI:10.32604/biocell.2021.014004
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.014004 | www.techscience.com/journal/biocell |
| Article |
POM analysis and computational interactions of 8-hydroxydiospyrin inside active site of protein tyrosine phosphatase 1B
1Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Umm Al-Qura University, Makkah, 21955, Saudi Arabia
2Department of Medicine Khyber Teaching Hospital, Peshawar, 25000, Pakistan
3Department of Chemistry, University of Swabi, Swabi, 23430, Pakistan
4LCM Laboratory, Mohammed First University, Faculty of Sciences, Oujda, 60000, Morocco
5Biology Department, Faculty of Sciences, University of Tabuk, Tabuk, 1144, Saudi Arabia
6Department of Biology, Faculty of Science, Ibb University, Ibb, Yemen
7Department of Chemistry, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, 22060, Pakistan
8Department of Pharmacy, University of Peshawar, Peshawar, 25120, Pakistan
9Department of Biotechnology, Shaheed Benazir Bhutto University, Sheringal, 14400, Pakistan
10Department of Pharmacy, Southeast University, Dhaka, 1213, Bangladesh
11Pharmakon Neuroscience Research Network, Dhaka, 1207, Bangladesh
12Department of Biochemistry, Abubakar Tafawa Balewa University, Bauchi, 740272, Nigeria
13Moscow State University of Technologies and Management (The First Cossack University), Moscow, 119991, Russian Federation
*Address correspondence to: Abdur Rauf, mashaljcs@yahoo.com
Received: 28 August 2020; Accepted: 27 November 2020
Abstract: Protein tyrosine phosphatase 1B (PTP1B) inhibition is considered as a potential therapeutic for the treatment of cancer, type 2 diabetes, and obesity. In our present work, we investigated the anti-diabetic potential of 8-hydroxydiospyrin (8-HDN) from D. lotus against the PTP1B enzyme. It showed significant inhibitory activity of PTP1B with an IC50 value of 18.37 ± 0.02 μM. A detailed molecular docking study was carried out to analyze the binding orientation, binding energy, and mechanism of inhibition. A comparative investigation of 8-HDN in the catalytic, as well as the allosteric site of PTP1B, was performed. Binding energy data showed that compound 8-HDN is more selective for the allosteric site and hence avoids the problems associated with catalytic site inhibition. The inhibition mechanism of 8-HDN can be further investigated as an active lead compound against PTP1B by using in vitro and in vivo models.
Keywords: Diospyros lotus; Roots; 8-Hydroxydiospyrin; Molecular docking; Protein tyrosine phosphatase 1B
Abbreviations
| 8-HDN: | 8-hydroxydiospyrin |
| PTP1B: | Protein tyrosine phosphatase 1B |
| T2D: | Type 2 diabetes |
Native to the tropics, Diospyros, known as data plum, is a genus of shrubs and evergreen trees. About 500 forms of the plant are known globally, of which 24 species are mostly found in India (Uddin et al., 2011a). The importance of different species might refer to either as their dark timber called ebony trees or their fruit called persimmon trees. Diospyros lotus L. (Ebenaceae), a deciduous tree, is extensively cultivated in tropical zones of Asia and Southeast Europe due to its resistance to drought. Its fruit has been shown to have anti-tumor and anti-diabetic competency (Hamedia and Shojaosadati, 2019), antiseptic and febrifuge, as well as a medicating agent of constipation (Rauf et al., 2015). In addition, a number of papers have highlighted the various applications of D. lotus including its nutritional content (Glew et al., 2005), being employed as medical agents (Loizzo et al., 2009; Rauf et al., 2017; Rauf et al., 2015), antidiarrheal activity (Rauf et al., 2014), mitigating oxidative stress (OS) through scavenging free radicals (Rauf et al., 2017), protecting cisplatin-induced OS (Cho et al., 2016; Saral et al., 2016), pro-inflammatory mediators (Cho et al., 2016). Protein tyrosine phosphates are referred to as a diverse family of enzymes that mostly disaccord the regular activities accomplished by protein tyrosine kinases (Gurzov et al., 2015). Several recent studies revealed that the PTP enzyme plays critical roles in signaling pathways. In this light, controlling the level of protein tyrosine phosphorylation is considered as a rampant mechanism that involves fundamentally in intracellular activities such as transcription, differentiation, and migration (Tonks, 2006; Nagata et al., 2012; Li et al., 2013). Among PTP family members, PTP1B is found in the cell and it is a type of PTP that is not associated with receptors. It is an interesting target for several disorders such as obesity and type 2 diabetes (T2D) (Kennedy and Ramachandran, 2000). Also, PTP1B contributes to the negative regulation of leptin- and insulin-receptor as reported by some genetic and biochemical studies (Koren and Fantus, 2007). In support of this, elevated insulin sensitivity, increased glycemic regulation, and resistance to obesity induced by diet were recorded in PTP1B-knockout experimental mice (Ali et al., 2009). Thus, PTP1B inhibition can serve as a novel target in the control of obesity and type 2 diabetes mellitus. Consistently, scientists have intensified their efforts to isolate novel and natural PTP1B inhibitors globally.Therefore, in this study, 8-hydroxydiospyrin (8-HDN) (Fig. 1) from D. lotus was screened for PTP1B inhibiting activity. By molecular docking model, to display the molecular interaction between PTP1B and the 8-HDN.
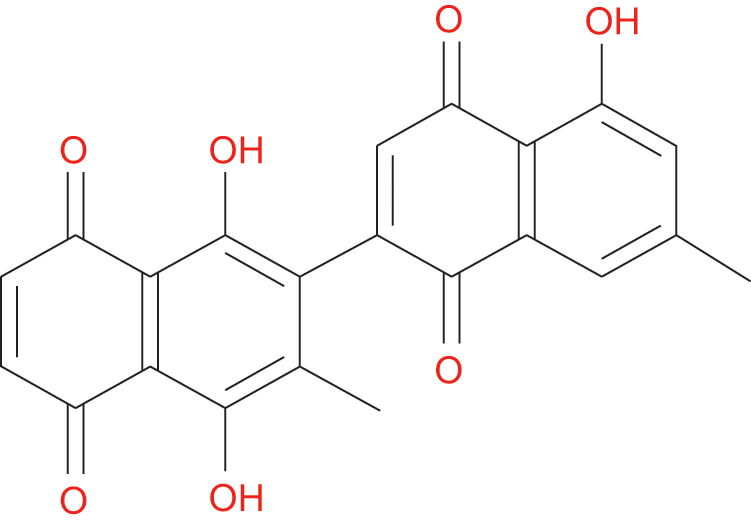
Figure 1: Chemical structure of 8-HDN.
Plant material (Diospyros lotus)
Diospyros lotus L. roots were obtained from Toormang Razagram, Pakistan. The root samples of the plant were authenticated by Dr. Abdur Rashid of the Department of Botany, University of Peshawar, Pakistan. The voucher specimen number, RF/01, was deposited at the Herbarium of the institution.
Extraction of the plant sample and isolation of the compound
The air-dried root samples of D. lotus were pulverized using an electric blender, after which 14 kg of the pulverized sample was weighted into another container. MeOH was added to the container containing the pulverized root sample of the plant and allowed to stand for 6 days. Thereafter, the mixture of the plant with the MeOH was filtrated and the filtrate was concentrated through the process of evaporation using a rotary vacuum evaporator at 50°C and reduced pressure. After the extraction process, the extract obtained was weighed (202 g) was defatted with hexane to remove color and dyes. The crude extract was subjected to various solvents to obtain various fractions such as hexane, chloroform, and ethyl acetate. The chloroform fraction (20 g) was subjected to repeat normal phase column chromatographically, which afforded 8-HDN (1.24 g).
The enzyme (PTP1B) inhibition analysis was performed in 96-well plates in the presence of 3,3-dimethyl glutarate buffer, pH = 7.0. The reaction mixture includes p-nitrophenol phosphate (pNPP) at a concentration of 1 mM, PTB1B at a concentration of 10 mM, and various concentrations of 8-HDN as per our recently published method (Bawazeer et al., 2019). Following the incubation period at 27°C for 40 min, the absorbance at 405 nm of the released pNPP was recorded. The procedure was performed in triplicates, and IC50 values were evaluated.
In this study, Molecular Operating Environment software, (version 2016.0802) was used to dock 8-HDN (Alhumaydhi et al., 2021). The three-dimensional (3-D) structure of enzyme PTP1B in complex with catalytic inhibitor was retrieved from protein data Bank (PDB ID = 1NNY). While the 3-D crystal structure of PTP1B with allosteric inhibitor was obtained from PDB (ID = 1T49). Preparation of ligands (ursolic acid and 8-HDN) and downloaded proteins (3D protonation, energy minimization, and determination of binding site were carried out by our previously reported methods (Jan et al., 2020; Tanoli et al., 2019; Iftikhar et al., 2018). All the ligand structures were drawn using the Builder option in MOE. A database of compounds was built as ligand.mdb. The compounds were then energy minimized up to 0.001 Gradient using MMFF94X forcefield. The enzyme structure was opened in the MOE window. The 3D protonation was done for all atoms in an implicit solvated environment at pH = 7, temperature = 300 K, and salt concentration of 0.1. The complete structure was energy minimized using MMFF94X forcefield. Finally, all the compounds were docked into the binding sites of the prepared enzymes. Default docking parameters were set, and ten different conformations were generated for each compound. The lowest binding energy ligand enzyme complexes were analyzed by the MOE ligand interaction module. While, for the 3-D interaction plot, a discovery studio visualizer was used (Biovia Systems 2017). While the surface model was created using Chimera (2020-09-08) (Pettersen et al., 2004)
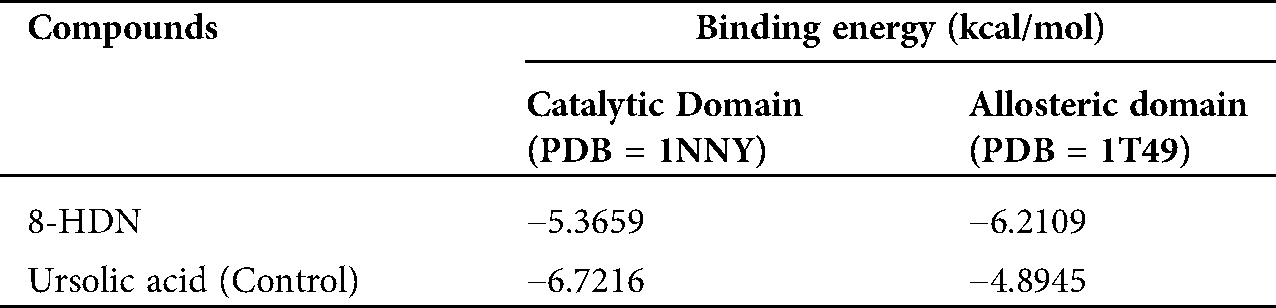
The chemical structure of 8-HDN was characterized by using physical and spectroscopic data recently published by our group (Uddin et al., 2013; Ullah et al., 2015). The chloroform fraction (20 g) was subjected to repeat normal phase column chromatographically, which afforded 8-HDN (1.24 g). The inhibitory activity of the compound (8-HDN) on PTP1B gave an IC50 value of 18.37 ± 0.02 μM. The IC50 value was greater than the value obtained for ursolic acid (control), which has an IC50 value of 3.21 ± 0.02 μM (Tab. 1).
Table 1: Protein tyrosine phosphatase 1B (PTP1B) inhibition activity of 8-HDN and ursolic acid

We have performed a detailed molecular docking study to analyze the binding orientation, binding energy, and mechanism of inhibition. For the mode of inhibition, we carried out a comparative investigation of 8-HDN in catalytic as well as the allosteric site of protein tyrosine phosphatase 1B (PTP1B). It has been reported in the literature that the inhibition of the catalytic domain could result in off-target undesirable side effects. While the allosteric site is not well conserved among phosphatases and hence avoids the problems associated with catalytic site inhibition. For the current study, the 3-D structure of enzyme PTP1B in complex with catalytic inhibitor was retrieved from protein data Bank (PDB ID = 1NNY). While the 3-D crystal structure of PTP1B with allosteric inhibitor was obtained from PDB (ID = 1T49). Ribbon and surface superimposed models of the two retrieved proteins are shown in Figs. 2a and 2b. The catalytic site is centered at Cys215. It includes a WPD loop (Trp179, Pro180, and Asp181). While the allosteric site is located nearly 20 Å away from Cys215 (Fig. 2a).
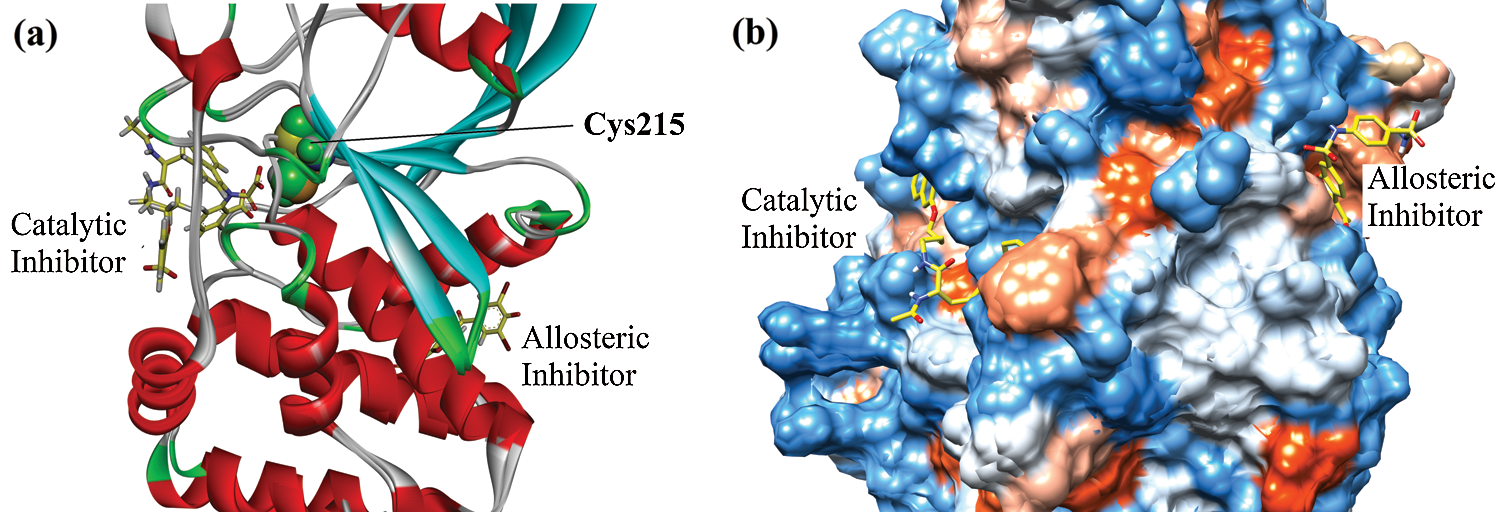
Figure 2: Superimposed ribbon (a) and surface (b) diagram of natives into the catalytic and allosteric binding site of protein tyrosine phosphatase 1B (PTP1B).
Before docking of 8HDN and control (ursolic acid), we validated the docking protocol by using the redock method. The computed root-mean-square deviation (RMSD) for redocking of ligands from both studied proteins showed the reliability of the docking algorithm (RMSD for 1NNY = 0.86 Å; RMSD for 1T49 = 0.93 Å). The superimposed ribbon and surface diagram of 8-HDN and native catalytic/allosteric site inhibitors are shown in Figs. 3a and 3b. Three-dimensional interaction plot into the binding site of catalytic site (PDB ID INNY) revealed that the compound under study forms three hydrogen bond interactions with Arg24, Arg254 and Gln262. Met258 forms π-sulfur interactions. A weak π-alkyl interaction also helps to stabilize the ligand-enzyme complex (Fig. 4a). A two-dimensional (2-D) interaction plot of the compound into the catalytic site is shown in Fig. 4b. The computed binding energy for compound 8-HDN in the catalytic site is −5.3659 kcal/mol.
A three-dimensional interaction plot into the allosteric binding site (ID = 1T49) revealed that the affinity of the compound is favored by three hydrogen bonds and five hydrophobic interactions. Ser187 and Asn193 form hydrogen bond interactions with carbonyl oxygen. While Glu276 forms hydrogen bond interactions with the hydroxyl group. Leu192, Phe196 and Phe280 forms π-π stacking interactions with 5,8-dihydronaphthalen rings (Fig. 5a). 2-D interaction plot of the compound into the allosteric site is shown in Fig. 5b. The computed binding energy for compound 8-HDN in the allosteric site is −6.2109 kcal/mol. While for control (Ursolic acid) is −4.8945 kcal/mol (Tab. 2).
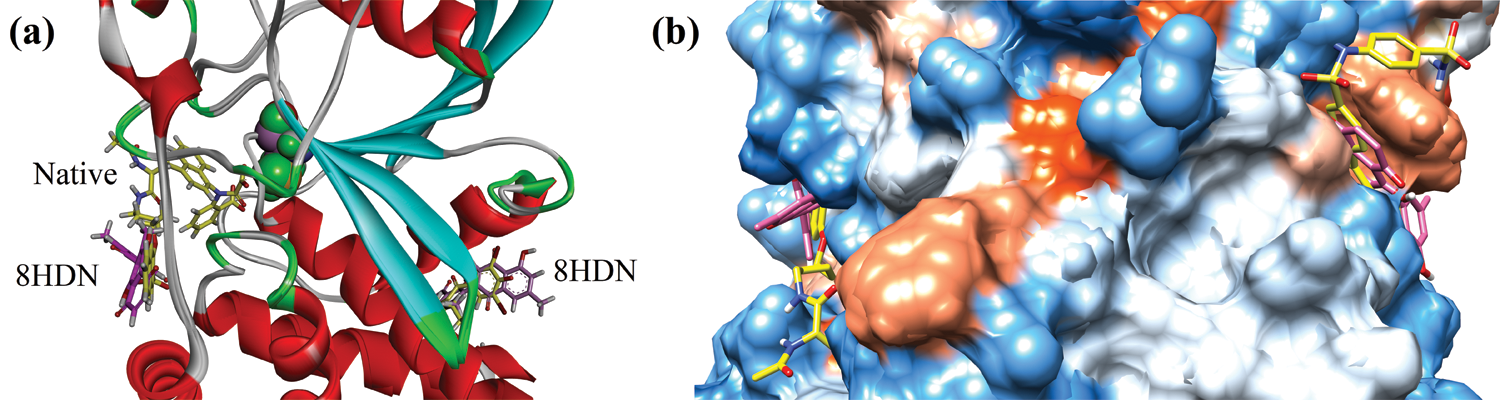
Figure 3: Superimposed ribbon (a) and surface diagram of compound 8-HDN (pink stick) and natives (yellow) into the catalytic and allosteric binding site.
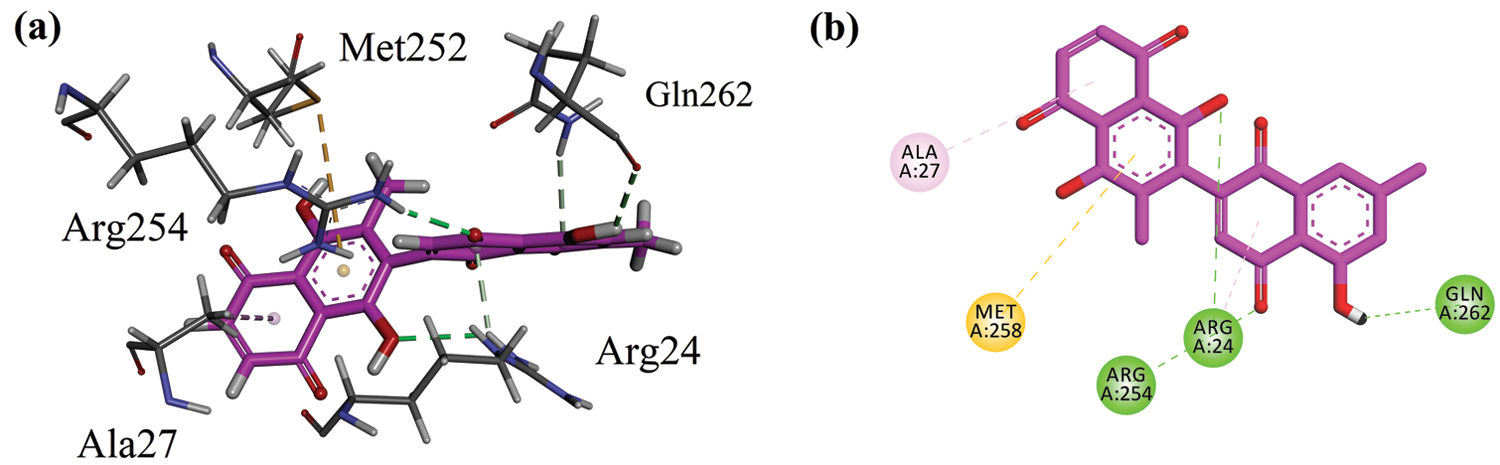
Figure 4: (a) Close-up 3-D interaction plot of the compound 8-HDN into the catalytic binding site of protein tyrosine phosphatase 1B (PTP1B, PDB ID = 1NNY) (b) 2-D interaction plot.
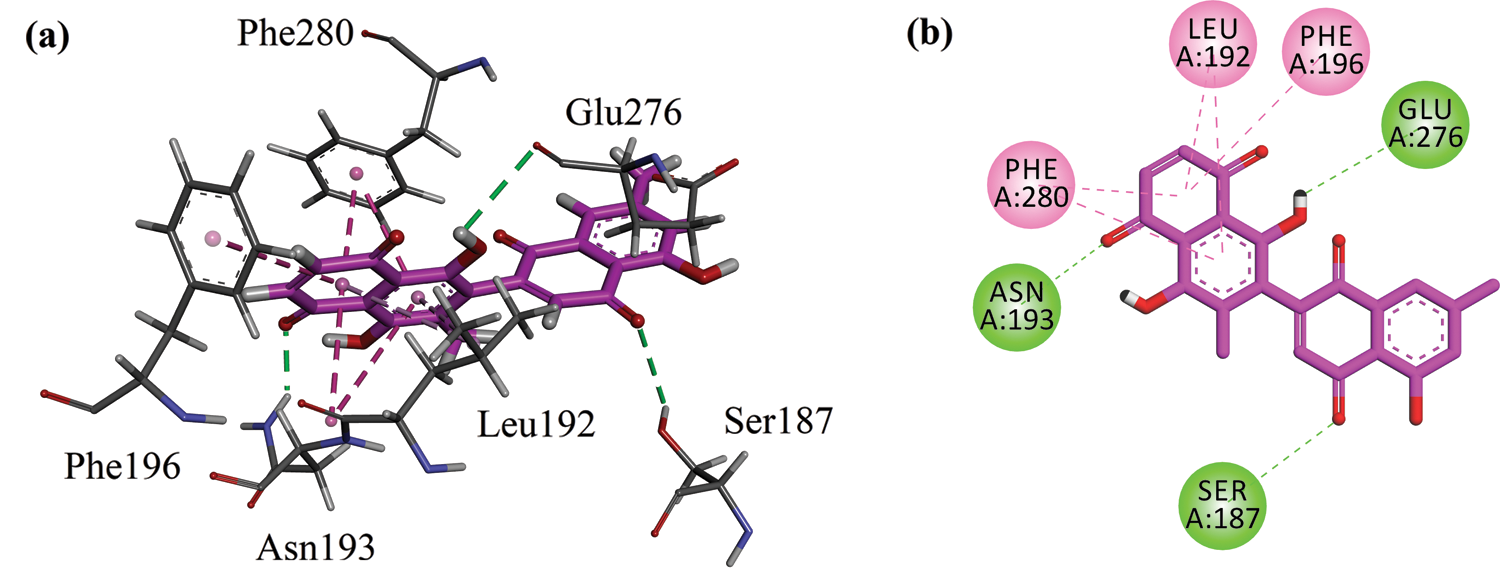
Figure 5: (a) Close-up 3-D interaction plot of the compound 8-HDN into the allosteric binding site of protein tyrosine phosphatase 1B (PTP1B, PDB ID = 1T49) (b) 2-D interaction plot.
The binding energy computed for ursolic acid (positive control) in the catalytic site is −6.7216 kcal/mol (Tab. 1). It forms four hydrogen bond interactions with Arg24, Arg254, and Gly259 (Figs. 6a and 6b).
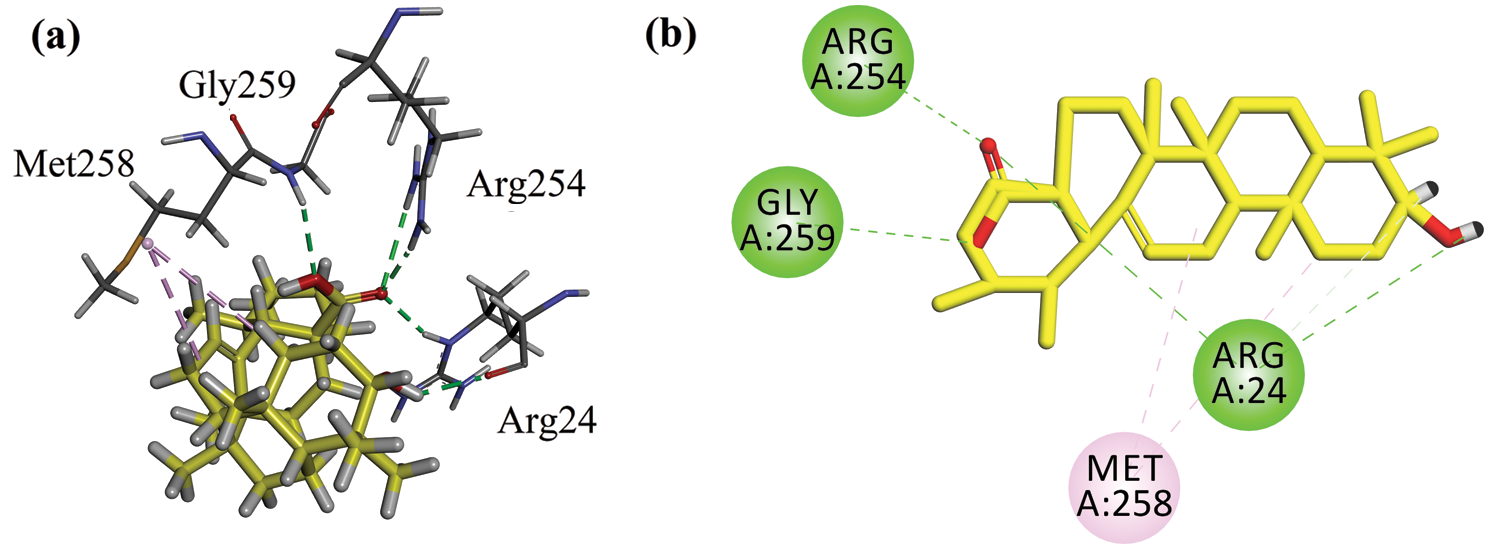
Figure 6: (a) Close-up 3-D interaction plot of the compound ursolic acid (positive control) into the catalytic binding site of protein tyrosine phosphatase 1B (PTP1B, PDB ID = 1NNY) (b) 2-D interaction plot.
Table 2: Binding energy values (in kcal/mol) computed via MOE docking into the binding site of catalytic and allosteric domains of PTP1B
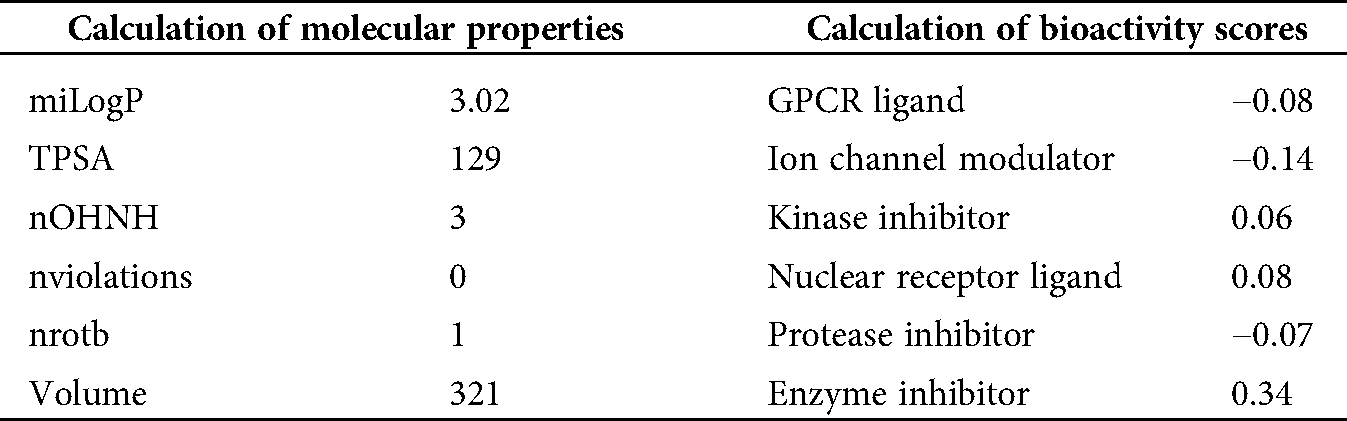
A potential drug candidate must have a good pharmacological profile with pharmacokinetic properties. Among in silico prediction tools. Petra, Osiris and Molinspiration (POM) calculations have been developed and documented for years to access the pharmacokinetic profile (Hakkou et al., 2017; Mabkhot et al., 2016; Rauf et al., 2015; Tighadouni et al., 2016; Sajid et al., 2016; Abdelhady et al., 2015; Header et al., 2015; Ben Hadda, 2015) to form sets of pharmacologically and diverse important conformers and tautomer, which can be used within 2D pharmacophore search procedures to elevate the number of meaningful hits of such test. These POM analyses give some information about the general limitations in the area of 2D structure and conformers/tautomer generation. The results of POM calculations are briefly described and discussed, and some outcomes obtained with the different tools are given. The results of the analysis are shown in Tab. 3.
Table 3: Molinspiration calculations of molecular properties and bioactivity scores of 8-HDN
Current research indicates the promising potential of 8-HDN to be further explored and developed as a novel compound targeting PTP1B, especially in diabetes and cancer. Recently, enzyme inhibitory activity of similar compound diospyrin has been reported on DNA gyrase of Mycobacterium tuberculosis (Karkare et al., 2013), as well as anticancer and antiparasitic activities of its derivatives and analogs (Dev et al., 2012; Kumar et al., 2012).
The design or identification of protein tyrosine phosphatase 1B (PTP1B) is an attractive area of research for medicinal/drug discovery researchers. However, there are a few challenges in developing PTP1B inhibitors. The catalytic site and its surrounding sub-sites have highly conserved polar architecture resulting in low bioavailability and off-target side effects. A few strategies have been developed to address these challenges (Kumar et al., 2018; Wiesmann et al., 2004). Wiesmann et al. (2004) discovered a druggable and non-conserved allosteric pocket (20 Å away from the active site) formed by more hydrophobic Leu192, Phe196 and Phe280. Asn193, Glu276 and Trp291 of α3 and α6 helices also interact with the inhibitors. There are a number of studies reported in the literature about the inhibition mechanism of PTP1B. These studies were carried out via inhibition kinetics and docking simulations. In a study carried out by Na et al. (2007), they revealed that naturally occurring amentoflavone from Selaginella tamariscina showed allosteric inhibition of PTB1B. Cai et al. (2015) reported in-vitro inhibition of PTP1B and docking studies of fifteen identified constituents from Anoectochilus chapaensis. The IC50 values of the nine active compounds were found in the range of 1.16–6.21 μM. Docking studies were carried out on the catalytic site of 1NNY, and the computed binding energy values were found between −7.4 to −8.5 kcal/mol. The tested compounds showed interactions with catalytic domain residues. Recently, Mphahlele et al. (2020) presented in-vitro and docking studies of 5-acetyl-2-aryl-6-hydroxybenzo[b]furans. The IC50 values of the nine active compounds were found in the range of 11.9–31.88 μM. Mechanism of inhibition was also investigated catalytic (PDB = 1NNY) as well as the allosteric site (PDB = 1T49) of protein tyrosine phosphatase 1B (PTP1B) via docking simulations. The computed binding energy values were found between −5.35 to −7.81 kcal/mol for catalytic inhibition. While for allosteric inhibition, the binding energies range from −6.82 to −11.20 kcal/mol. They concluded that the studied compounds are more selective for the allosteric site. Paudel et al. (2018) evaluated the PTP1B inhibitory potential of three principal components: mulberrofuran G, albanol B, and kuwanon G in M. alba rootbark. The studied compounds showed allosteric PTP1B inhibition via Asn193 and Glu276. While for catalytic inhibition, their mode of inhibition was through Arg24, Tyr46, Asp48, and Arg254.
In the current study, we investigated the mechanism of PTP1B inhibition by 8-HDN. Catalytic site inhibition by 8-HDN with a binding energy value of −5.3659 kcal/mol established three hydrogen bond interactions, a π-sulfur interaction, and a weak π-alkyl interaction. While the computed binding energy for positive control was −6.7216 kcal/mol. The PTP1B complex with 8-HDN allosteric site showed a binding energy value of −6.2109 kcal/mol. Binding energy data showed that compound 8-HDN is more selective for the allosteric site.
Pi-charge calculation and molecular structure optimization
The charge repetition of 8-HDN shows an important combined antibacterial/antifungal and antiviral O, O, O-pharmacophore site (Figs. 7 and 8), which deserves a separate supplementary antiviral/antiparasite screening. Thus, we have started this compound, and other achievements can be made. Our previous experience with similar flavonoids molecules indicates that a subtle change in pharmacophore can lead us to more efficient antioxidant and antinociceptive and anti-inflammatory agents (Ben Hadda et al., 2013; Rauf et al., 2016).
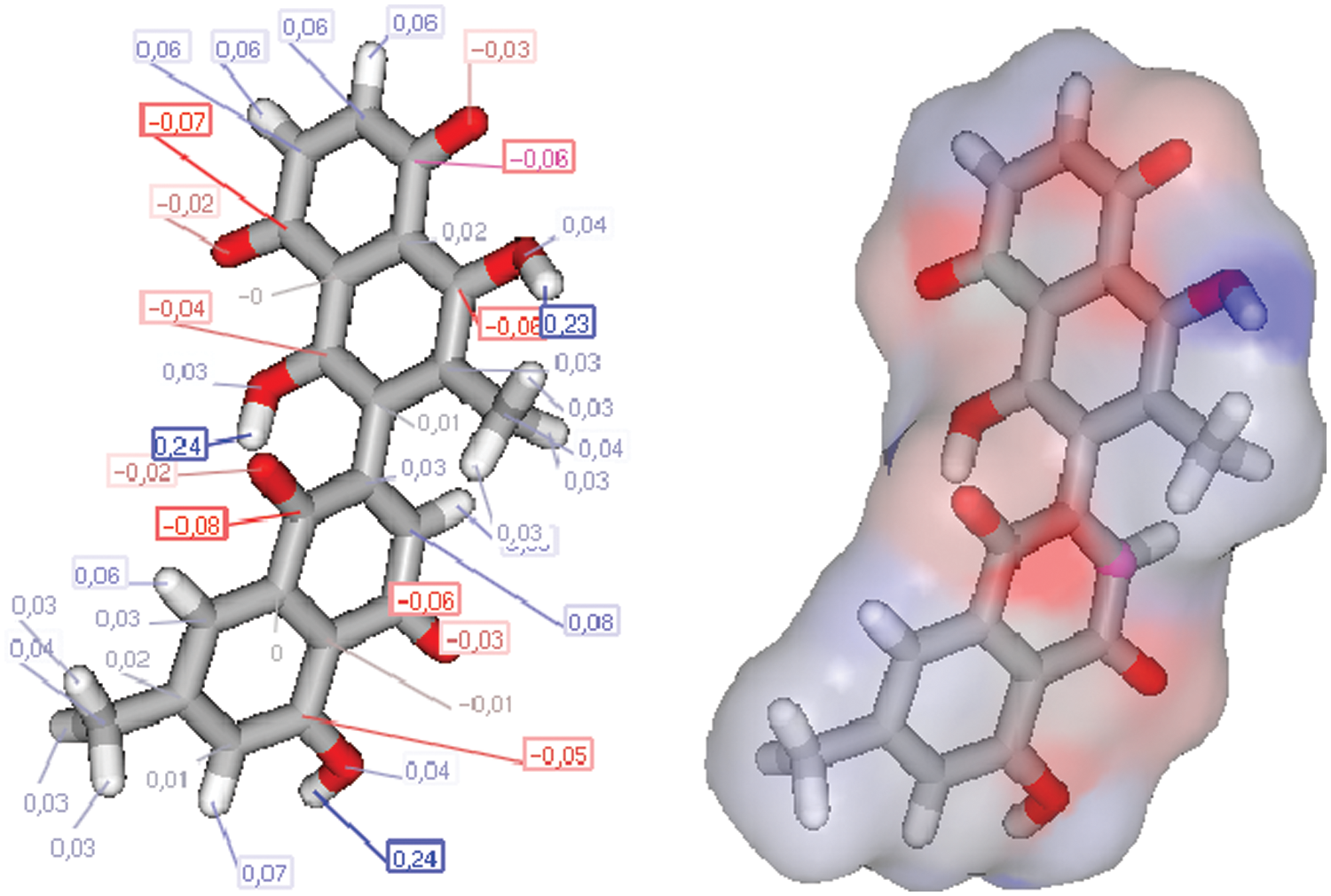
Figure 7: Charges and molecular structure optimization of 8-HDN.
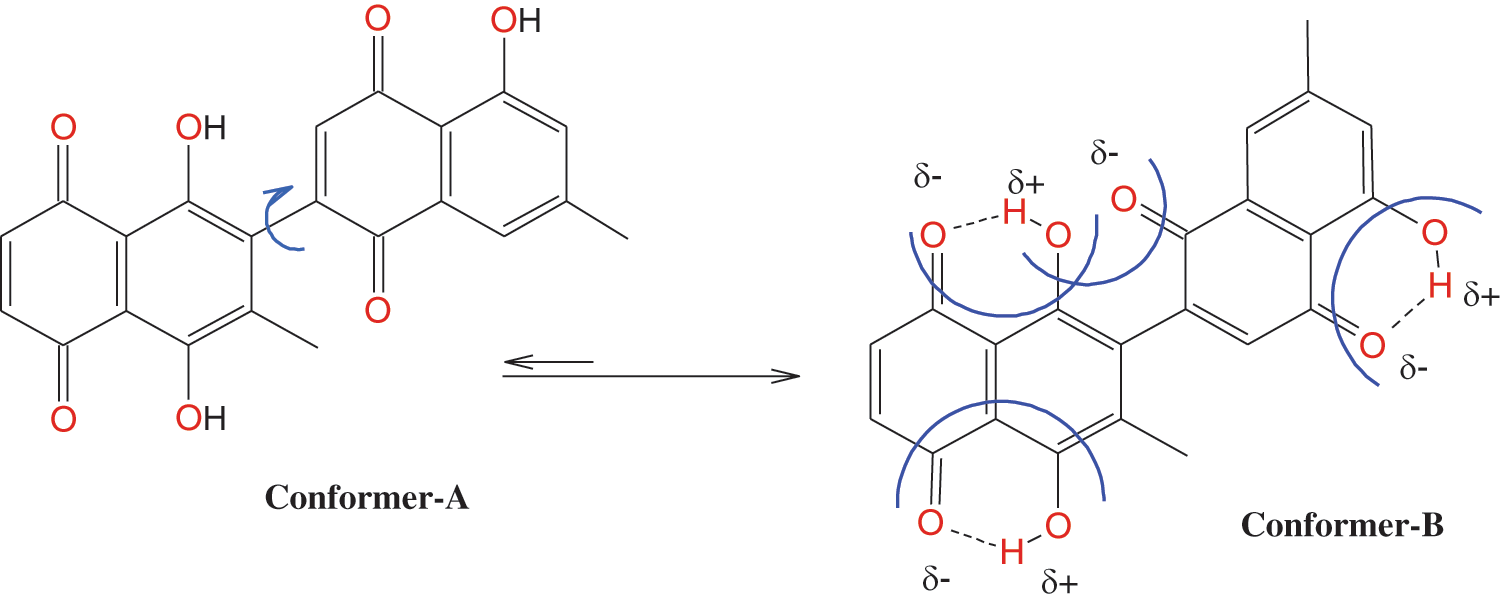
Figure 8: Conformation of 8-HDN.
The theoretical toxicity risks determination for the 8-HDN using the Osiris program indicated that this flavonoid (Fig. 9) causes fewer side effects compared to the standard clinical drugs. It also revealed that 8-HDN can serve as an antibiotic with some pharmacomodulation (DS = 0.52). From the data estimated in Fig. 9, the structure is not supposed to mutagenic when analyzed via the mutagenicity assessment of the molecular system. Based on the reproductive and irritating effects, 8-HDN is at low risk compared with the control. The hydrophilicity character of the compound has been shown in terms of the cLogP value. It has been established that the permeation or absorption is highly affected by the hydrophilicity (cLogP < 5).
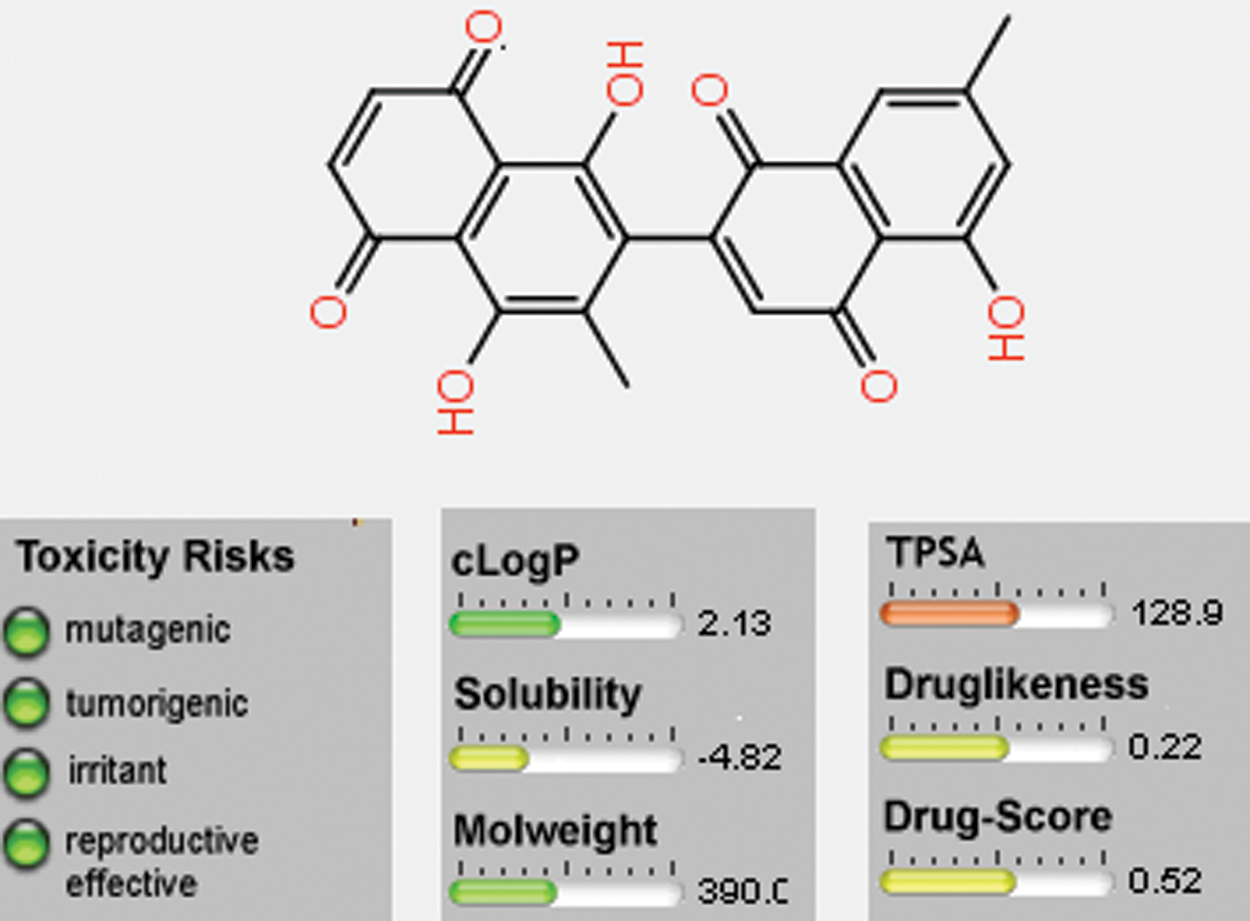
Figure 9: Osiris calculations of molecular properties of 8-HDN.
Petra, Osiris and Molinspiration (POM) analysis is a well-known bioinformatics tool to identify the pharmacophore sites and predict the biological activities of molecules on the basis of steric/electrostatic properties. According to Molinspiration calculation, when miLogP is greater than 5, the permeation or absorption reduces. As a result of this, the compound (8-HDN) has a miLogP value within the acceptable criteria, but other vital indices should be considered. This is linked to the geometrical configuration of the pharmacophore site (Fig. 5) because it is flexible for 8-HDN. We have calculated the molecular properties (TPSA, number of violations and volume) for the compound, and we have noted that they could be used as potential hits. Theoretical drug scores calculated via the online Molinspiration program are presented in Tab. 3. The calculation of bioactivity scores combines ion channel modulator, nuclear receptor ligand, kinase inhibitor, GPCR ligand, enzyme inhibitor and protease inhibitor in five separate values that may be employed to investigate the 8-HDN’s total ability to qualify as a drug. The studied compound showed a promising ability to act as an enzyme inhibitor (Tab. 2).
In conclusion, to the best of our knowledge, no report is available on the antidiabetic potential of hydroxydiospyrin and D. lotus; our results deserve attention. Further studies, based on in vivo models, are needed to further elucidate this relevant biological activity. Docking studies were carried out to investigate the mechanism of inhibition. Binding orientation and binding energy data were computed into the catalytic (−5.3659 kcal/mol) and allosteric (−6.2109 kcal/mol) binding site of PTP1B. Binding energy data showed that compound 8-HDN is more selective for the allosteric site. 8-HDN can be further screened as an active lead compound against PTP1B by using in vitro and in vivo models. Overall, we found that 8-HDN structure optimizations based on the performed POM analysis.
Acknowledgement: The authors are highly thankful to the Higher Education commission, Pakistan for funding this research group No. NRPU649.
Authors Contributions: The authors confirm contribution to the paper as follows: Study conception and design: S. Bawazer, A. Khan, A. Rauf; data collection: T. B. Hadda, Y. S. Al-awthan; analysis and interpretation of results: O. Bahattab, U. Rashid, I. Khan; M. A. Nawaz draft manuscript preparation: M. S. Uddin, O. Ahmed, M. A. Shariati. All authors reviewed the results and approved the final version of the manuscript.
Availability of Data and Materials: The data including complete spectroscopic data of 8-hydroxydiospyrin (8-HDN) associated materials used to support the Research of this study are available from the corresponding authors upon request.
Funding Statement: This research was funded by Higher Education commission, Pakistan (HEC), Grant No. NRPU649.
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Abdelhady MIS, Kamal AM, Rauf A, Mubarak MS, Ben Hadda T. (2015). Bioassay-guided isolation and POM analyses of a new immunomodulatory polyphenolic constituent from Callistemon viridiflorus. Natural Product Research 30: 1131–1135. DOI 10.1080/14786419.2015.1045508. [Google Scholar] [CrossRef]
Ali MI, Ketsawatsomkron P, Chantemele EJ. (2009). Deletion of protein tyrosine phosphatase 1b improves peripheral insulin resistance and vascular function in obese, leptin-resistant mice via reduced oxidant tone. Circulation Research 105: 1013–1022. DOI 10.1161/CIRCRESAHA.109.206318. [Google Scholar] [CrossRef]
Alhumaydhi FA, Aljohani ASM, Rashid U, Shah ZA, Rauf A et al. (2021). In vivo antinociceptive, muscle relaxant, sedative, and molecular docking studies of peshawaraquinone isolated from fernandoa adenophylla (Wall. ex G. Don) steenis. ACS Omega 6: 996–1002. [Google Scholar]
Bawazeer S, Rauf A, Shahidullah A, Mishra A P, Faraone I, Milella L, Ullah K, Uddin G, Khan I, Patel S, Shah ZA. (2019). Structural insights behind protein tyrosine phosphatase 1B inhibitory activity of diospyrin. Indian Journal of Pharmaceutical Sciences 81: 565–568. DOI 10.36468/pharmaceutical-sciences.546. [Google Scholar] [CrossRef]
Ben Hadda T. (2015). Computational POM and DFT evaluation of experimental in-vitro cancer inhibition of staurosporine-ruthenium (II) complexes: The power force of organometallics in drug design. Acta Chimica Slovenica 62: 679–688. DOI 10.17344/acsi.2015.1357. [Google Scholar] [CrossRef]
Ben Hadda T, Fergoug T, Warad I, Masand V, Sheikh J. (2013). POM as a quick bioinformatic platform to select flavonoids and their metabolites as potential and efficient HIV-1 integrase inhibitors. Research on Chemical Intermediates 39: 1227–1244. DOI 10.1007/s11164-012-0679-6. [Google Scholar] [CrossRef]
Cai J, Zhao L, Tao W. (2015). Potent protein tyrosine phosphatase 1B (PTP1B) inhibiting constituents from Anoectochilus chapaensis and molecular docking studies. Pharmaceutical Biology 53: 1030–1034. DOI 10.3109/13880209.2014.957781. [Google Scholar] [CrossRef]
Cho BO, Yin HH, Park SH, Byun EB, Ha HY, Jang SI. (2016). Anti-inflammatory activity of myricetin from Diospyros lotus through suppression of NF-kB and STAT1 activation and Nrf2-mediated HO-1 induction in lipopolysaccharide- stimulated RAW264.7 macrophages. Bioscience, Biotechnology and Biochemistry 80: 1520–1530. DOI 10.1080/09168451.2016.1171697. [Google Scholar] [CrossRef]
Dev MA, Rajarajeshwari N, Ganapaty S. (2012). Antiprotozoal and anthelmintic naphthoquinones from three unexplored species of Diospyros. Journal of Natural Remedies 12: 129–134. [Google Scholar]
Glew RH, Ayaz FA, Millson M, Huang HS, Chuang LT, Sanz C, Golding JB. (2005). Changes in sugars, acids and fatty acids in naturally parthenocarpic date plum persimmon (Diospyros lotus L.) fruit during maturation and ripening. European Food Research and Technology 221: 113–118. DOI 10.1007/s00217-005-1201-9. [Google Scholar] [CrossRef]
Gurzov EN, Stanley WJ, Brodnicki TC, Thomas HE. (2015). Protein tyrosine phosphatases: Molecular switches in metabolism and diabetes. Trends in Endocrinology & Metabolism 26: 30–33. DOI 10.1016/j.tem.2014.10.004. [Google Scholar] [CrossRef]
Hakkou Z, Maciuk A, Leblais V, Bouanani N E, Mekhfi H, Bnouham M, Aziz M, Ziyyat A, Rauf A, Hadda TB, Shaheen U, Patel S, Fischmeister R, Legssyer A. (2017). Antihypertensive and vasodilator effects of methanolic extract of Inula viscosa: Biological evaluation and POM analysis of cynarin, chlorogenic acid as potential hypertensive. Biomedicine & Pharmacotherapy 93: 62–69. DOI 10.1016/j.biopha.2017.06.015. [Google Scholar] [CrossRef]
Hamedia S, Shojaosadati SA. (2019). Rapid and green synthesis of silver nanoparticles using Diospyros lotus extract: Evaluation of their biological and catalytic activities. Polyhedron 171: 172–1180. DOI 10.1016/j.poly.2019.07.010. [Google Scholar] [CrossRef]
Header E, ElSawy N, El-Boshy M, Basalamah M, Mubarak MS, Ben Hadda T. (2015). POM analyses of constituents of Rosmarinus officinalis and their synergistic effect in experimental diabetic rats. Journal of Bioanalysis & Biomedicine 15: 018–023. [Google Scholar]
Iftikhar F, Yaqoob F, Tabassum N, Jan MS, Sadiq A, Tahir S, Batool T, Niaz B, Ansari FL, Choudhary MI, Rashid U. (2018). Design, synthesis, in-vitro thymidine phosphorylase inhibition, in-vivo antiangiogenic and in-silico studies of C-6 substituted dihydropyrimidines. Bioorganic Chemistry 80: 99–111. DOI 10.1016/j.bioorg.2018.05.026. [Google Scholar] [CrossRef]
Jan M S, Ahmad S, Hussain F, Ahmad A, Mahmood F, Rashid U, Abid OUR, Ullah F, Ayaz M, Sadiq A. (2020). Design, synthesis, in-vitro, in-vivo and in-silico studies of pyrrolidine-2,5-dione derivatives as multitarget anti-inflammatory agents. European Journal of Medicinal Chemistry 186: 111863. DOI 10.1016/j.ejmech.2019.111863. [Google Scholar] [CrossRef]
Karkare S, Chung TT, Collin F. (2013). The naphthoquinone diospyrin is an inhibitor of DNA gyrase with a novel mechanism of action. Journal of Biological Chemistry 288: 5149–5156. DOI 10.1074/jbc.M112.419069. [Google Scholar] [CrossRef]
Kennedy BP, Ramachandran C. (2000). Protein tyrosine phosphatase-1B in diabetes. Biochemical Pharmacology 60: 877–883. DOI 10.1016/S0006-2952(00)00305-1. [Google Scholar] [CrossRef]
Koren S, Fantus IG. (2007). Inhibition of the protein tyrosine phosphatase PTP1B: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Practice & Research: Clinical Endocrinology & Metabolism 21: 621–640. DOI 10.1016/j.beem.2007.08.004. [Google Scholar] [CrossRef]
Kumar AP, Nguyen MN, Verma C, Lukman S. (2018). Structural analysis of protein tyrosine phosphatase 1B reveals potentially druggable allosteric binding sites. Proteins-Structure Function and Bioinformatics 86: 301–321. DOI 10.1002/prot.25440. [Google Scholar] [CrossRef]
Kumar B, Kumar A, Ghosh S. (2012). Diospyrin derivative, an anticancer quinonoid, regulates apoptosis at endoplasmic reticulum as well as mitochondria by modulating cytosolic calcium in human breast carcinoma cells. Biochemical and Biophysical Research Communications 417: 903–909. DOI 10.1016/j.bbrc.2011.12.072. [Google Scholar] [CrossRef]
Li X, Wilmanns M, Thornton J, Kohn M. (2013). Elucidating human phosphatase-substrate networks. Science Signaling 6: 10. DOI 10.1126/scisignal.6306er10. [Google Scholar] [CrossRef]
Loizzo MR, Said A, Tundis R, Hawas UW, Rashed K, Menichini F, Frega NG, Menichini F. (2009). Antioxidant and antiproliferative activity of Diospyros lotus L. extract and isolated compounds. Plant Foods for Human Nutrition 64: 264–270. DOI 10.1007/s11130-009-0133-0. [Google Scholar] [CrossRef]
Mabkhot YN, Arfan M, Zgou H, Genc ZK, Genc M, Rauf A, Bawazeer S, Ben Hadda T. (2016). How to improve antifungal bioactivity: POM and DFT study of some chiral amides derivatives of diacetyl-l-tartaric acid and amines. Research on Chemical Intermediates 42: 8055–8068. DOI 10.1007/s11164-016-2578-8. [Google Scholar] [CrossRef]
Mphahlele MJ, Choong YS, Maluleka MM, Gildenhuys S. (2020). Synthesis, in vitro evaluation and molecular docking of the 5-acetyl-2-aryl-6-hydroxybenzo [b] furans against multiple targets linked to type 2 diabetes. Biomolecules 10: 418. DOI 10.3390/biom10030418. [Google Scholar] [CrossRef]
Na M, Kim KA, Oh H, Kim BY, Oh WK, Ahn JS. (2007). Protein tyrosine phosphatase 1B inhibitory activity of amentoflavone and its cellular effect on tyrosine phosphorylation of insulin receptors. Biological and Pharmaceutical Bulletin 30: 379–381. DOI 10.1248/bpb.30.379. [Google Scholar] [CrossRef]
Nagata N, Matsuo K, Bettaieb A, Bakke J, Matsuo I, Graham J, Xi Y, Liu S, Tomilov A, Tomilova N, Gray S, Jung DY, Ramsey JJ, Kim JK, Cortopassi G, Havel PJ, Haj FG. (2012). Hepatic Src homology phosphatase 2 regulates energy balance in mice. Endocrinology 153: 3158–3169. DOI 10.1210/en.2012-1406. [Google Scholar] [CrossRef]
Paudel P, Yu T, Seong SH, Kuk EB, Jung HA, Choi JS. (2018). Protein tyrosine phosphatase 1B inhibition and glucose uptake potentials of mulberrofuran G, albanol B, and kuwanon G from root bark of Morus alba L. in insulin-resistant HepG2 cells: An in vitro and in silico study. International Journal of Molecular Sciences 19: 1542. DOI 10.3390/ijms19051542. [Google Scholar] [CrossRef]
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. (2004). UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry 25: 1605–1612. DOI 10.1002/jcc.20084. [Google Scholar] [CrossRef]
Rauf A, Uddin G, Siddiqui BS, Muhammad N. (2014). Antipyretic and antinociceptive activity of Diospyros lotus L. in animals. Asian Pacific Journal of Tropical Biomedicine 41: S382–386. [Google Scholar]
Rauf A, Uddin G, Siddiqui BS, Khan H, Ur-Rehman M, Warad I, Ben Hadda T, Patel S, Khan A, Farooq U. (2015). POM analysis of phytotoxic agents from Pistacia integerrima Stewart. Current Bioactive Compounds 11: 231–238. [Google Scholar]
Rauf A, Uddin G, Siddiqui BS, Khan H, Shah SUA, HaddaTB, MabkhotYN, Farooq U, Khan A. (2016). Antinociceptive and anti-inflammatory activities of flavonoids isolated from Pistacia integerrima galls. Complementary Therapies in Medicine 25: 132–138. DOI 10.1016/j.ctim.2016.02.002. [Google Scholar] [CrossRef]
Rauf A, Uddin G, Patel S, Khan A, Halim SA, Bawazeer S, Ahmad K, Muhammad N, Mubarak MS. (2017). Diospyros, an under-utilized, multi-purpose plant genus: A review. Biomedicine & Pharmacotherapy 91: 714–730. [Google Scholar]
Sajid Z, Ahmad M, Aslam S, Ashfaq UA, Zahoor AF, Saddique FA, Parvez M, Hameed A, Sultan S, Zgou H, Hadda TB. (2016). Novel armed pyrazolobenzothiazine derivatives: Synthesis, X-ray crystal structure and POM analyses of biological activity against drug resistant clinical isolate of Staphylococcus aureus. Pharmaceutical Chemistry Journal 50: 172–180. DOI 10.1007/s11094-016-1417-y. [Google Scholar] [CrossRef]
Saral S, Ozcelik E, Cetin A, Saral O, Basak N, Aydın M, Ciftci O. (2016). Protective role of Diospyros lotus on cisplatin-induced changes in sperm characteristics, testicular damage and oxidative stress in rats. Andrologia 48: 308–317. DOI 10.1111/and.12448. [Google Scholar] [CrossRef]
Tanoli ST, Ramzan M, Hassan A, Sadiq A, Jan MS, Khan FA, Ullah F, Ahmad H, Bibi M, Mahmood T, Rashid U. (2019). Design, synthesis and bioevaluation of tricyclic fused ring system as dual binding site acetylcholinesterase inhibitors. Bioorganic Chemistry 83: 336–347. DOI 10.1016/j.bioorg.2018.10.035. [Google Scholar] [CrossRef]
Tighadouni S, Radi S, Sirajuddin M, Akkurt M, Özdemir N, Ahmad M, Mabkhot YN, Ben Hadda T. (2016). In vitro antifungal, anticancer activities and POM analyses of a novel bioactive Schiff base 4-{[(E)-furan-2-ylmethylidene]amino}p-henol: Synthesis, characterization and crystal structure. Journal of the Chemical Society of Pakistan 38: 157–165. [Google Scholar]
Tonks NK. (2006). Protein tyrosine phosphatases: From genes, to function, to disease. Nature Reviews Molecular Cell Biology 7: 833–846. DOI 10.1038/nrm2039. [Google Scholar] [CrossRef]
Uddin G, Rauf A, Siddiqui BS (2011a). Preliminary comparative phytochemical screening of Diospyros lotus Stewart. Middle-East Journal of Scientific Research 10: 78–81. [Google Scholar]
Uddin G, Rauf A, Arfan M. (2013). Molecular docking of diospyrin as a LOX inhibitory compound. Journal of Saudi Chemical Society 20: S448–450. DOI 10.1016/j.jscs.2013.01.009. [Google Scholar] [CrossRef]
Uddin G, Rauf A, Rehman TU (2011b). Phytochemical screening of Pistacia chinensis var. integerrima. Middle-East Journal of Scientific Research 7: 707–711. [Google Scholar]
Ullah Z, Ata-ur-Rahman, Fazl-i-Sattar, Rauf A, Yaseen M, Hassan W, Tariq M, Ayub K, Tahir AA, Ullah H. (2015). Density functional theory and phytochemical study of 8-hydroxyisodiospyrin. Journal of Molecular Structure 1095: 69–78. DOI 10.1016/j.molstruc.2015.04.027. [Google Scholar] [CrossRef]
Wiesmann C, Barr KJ, Kung J, Zhu J, Erlanson DA, Shen W, Fahr B, Zhong M, Taylor L, Randal M, McDowell RS, Hansen SK. (2004). Allosteric inhibition of protein tyrosine phosphatase 1B. Nature Structural & Molecular Biology 11: 730–737. DOI 10.1038/nsmb803. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |