DOI:10.32604/biocell.2021.014332
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.014332 | www.techscience.com/journal/biocell |
| Review |
Extraintestinal manifestations of inflammatory bowel disease, nitroxidative stress and dysbiosis: What is the link between them?
1Programa de Pós-Graduação da Rede Nordeste de Biotecnologia (RENORBIO), Universidade Federal de Alagoas (UFAL), Maceió, AL 57072-970, Brazil
2Programa de Pós-Graduação em Ciências da Saúde (PPGCS), Universidade Federal de Alagoas (UFAL), Maceió, AL 57072-970, Brazil
3Programa de Pós-Graduação em Química e Biotecnologia (PPGQB/UFAL), Campus A. C. Simões, Avenida Lourival Melo Mota, s/n, Tabuleiro dos Martins, Maceió, AL 57072-970, Brazil
4Programa de Pós-Graduação em Nutrição (PPGNU/UFAL), Programa de Pós-Graduação em Ciências Médicas (PPGCM/UFAL), Universidade Federal de Alagoas (UFAL), Maceió, AL 57072-970, Brazil
*Address correspondence to: Fabiana Andréa Moura, fabiana.moura@fanut.ufal.br
Received: 17 September 2020; Accepted: 20 November 2020
Abstract: Inflammatory bowel disease (IBD), which includes Crohn’s disease and ulcerative colitis, has a not yet completely defined aetiology and is characterized by a progressive chronic inflammation that involves nitroxidative stress and dysbiosis. Extraintestinal manifestations can occur and affect several organs, including the liver and bile ducts, joints, skin, eyes, and less frequently, the heart, brain, and kidneys, increasing the risk of morbidity and mortality. These repercussions may be associated with the activity or severity of IBD. The present review proposes to report and analyse the participation of dysbiosis and nitroxidative stress in the genesis of extraintestinal manifestations, aiming to contribute to a better understanding of the disease and to focus on the development of individualized preventive and therapeutic strategies.
Keywords: Oxidative stress; Microbiota; Bacterial translocation; Ulcerative colitis; Crohn disease
Abbreviations
| 4-HNE: | 4-hydroxy-2-nonenal |
| AP-1: | activating protein-1 |
| HLA-B27: | B27-human leukocyte antigen |
| BBB: | blood-brain barrier |
| BDNF: | brain-derived neurotrophic factor |
| BMD: | bone mineral density |
| CAT: | catalase |
| CCL25: | C-C motif chemokine ligand 25 |
| CNS: | central nervous system |
| CCR6: | chemokine receptors 6 |
| D161: | cluster of differentiation 161 |
| CRP: | C-reactive protein |
| CD: | crohn disease |
| CYP27A1: | cytochrome P450 family 27 subfamily A member 1 |
| CYP7A1: | cytochrome P450 family 7 subfamily A member 1 |
| EIMs: | extraintestinal manifestations |
| GPx: | glutathione peroxidase |
| HM: | hepatobiliary manifestations |
| HCy: | homocysteine |
| HLA-DR1: | human leukocyte antigen-DR1 |
| IgA: | immunoglobulin A |
| iNOS: | inducible nitric oxide synthase |
| IBD: | inflammatory bowel disease |
| ICAM-1: | intercellular Adhesion Molecule-1 |
| IFN-γ: | interferon gamma |
| IL: | interleukin |
| LRR: | leucine rich repeats |
| LPS: | lipopolysaccharide |
| MDA: | malondialdehyde |
| MAPK: | mitogen activated protein kinase |
| MCP-1: | monocyte chemoattractant protein-1 |
| MAdCAM-1: | mucosal vascular addressin cell adhesion molecule-1 |
| MAIT: | mucosal associated invariant T cells |
| MS: | multiple sclerosis |
| NAFLD: | non-alcoholic fatty liver disease |
| NFκB: | nuclear factor kappa B |
| NLRP3: | nucleotide-binding domain, leucine-rich-repeat-containing receptor 3 |
| NOD: | nucleotide-binding oligomerization domain |
| OmpC: | outer membrane protein C |
| oxLDL: | oxidized low density lipoprotein |
| PAMP: | pathogen-associated molecular patterns |
| PD: | parkinson disease |
| PPARs: | peroxisome Proliferator-Activated Receptors |
| PRR: | pattern recognition receptors |
| ONOO-: | peroxynitrite |
| PSC: | primary sclerosing cholangitis |
| RONS: | reactive Oxygen and Nitrogen Species |
| ROS: | reactive Oxygen Species |
| SCFA: | short chain fatty acids |
| STAT3: | signal transducer and activator of transcription 3 |
| SpA: | spondyloarthropathies |
| SREBP: | sterol Regulatory Element Binding Protein |
| TLR4: | toll like receptor 4 |
| TRAF3IP2: | tumor necrosis factor alfa receptor-associated factor interacting protein 2 |
| TMA: | trimethylamine |
| TMAO: | trimethylamine-N-oxide |
| TNF-α: | tumor necrosis factor alfa |
| UC: | ulcerative colitis |
| VCAM-1: | vascular adhesion molecule-1 |
| VAP-1: | vascular adhesion protein-1 |
| VEGF: | vascular endothelial growth factor. |
Inflammatory bowel disease (IBD) is a chronic and recurrent disease that includes Crohn’s disease (CD) and ulcerative colitis (UC). The IBD aetiology is not fully elucidated, however, it is known that there is an intimate interaction between genetic, immunological, and environmental factors (diet, smoking, circadian cycle), the intestinal microbiota and nitroxidative stress (Manichanh et al., 2012; Moura et al., 2016; Garber and Regueiro, 2019; Feuerstein and Cheifetz, 2014).
Intestinal microbiota comprises more than 150.000 species of commensal microorganisms that inhabit the gastrointestinal tract and perform beneficial functions to the host, such as the synthesis of substances important for energy metabolism, defense against luminal pathogens, and modulation of the immune response (Qin et al., 2010; Nishida et al., 2018).
The participation of the microbiota in the modulation of innate and adaptive immunity of the mucosa is fundamental to maintain the integrity of the epithelial barrier and occurs through the interaction between the pathogen-associated molecular patterns (PAMPs) and the pattern recognition receptors (PRR), such as Toll-like receptors (TLR), present in immune cells, as well as by the action of their metabolites, especially short-chain fatty acids (SCFA), which act to induce immunological tolerance by stimulating, mainly, the polarization of regulatory T lymphocytes.
In addition, they secrete or promote the secretion of antimicrobial factors, such as defensins and immunoglobulin A (IgA) (Kamada and Núñez, 2013; Hart et al., 2005; Nishida et al., 2018).
Imbalance in microbial diversity and density, known as dysbiosis, can alter the interaction between the host-microbiota-immune system and has been associated with the appearance of many inflammatory and autoimmune disorders, including IBD. Some studies have reported that IBD patients have changes in microbial composition, when compared to healthy individuals, with a decrease in SCFA producing commensal bacteria, such as Faecalibacterium prausnitzii, and an increase in mucolytic, sulfate-producing, and pathogenic bacteria (Fujimoto et al., 2013; Takahashi et al., 2016; Nishino et al., 2018; Nishida et al., 2018).
It has been described that the increase in intestinal permeability and the unregulated immune response causes a continuous inflammatory process (with neutrophilic infiltration) and a redox imbalance, generating nitroxidative stress, production of pro-inflammatory cytokines, and consequent impairment of the intestinal barrier, characterized by the destruction of tight junctions and oxidative damage caused by lipid peroxidation (Fig. 1) (Moura et al., 2015). This scenario allows pathogenic bacteria and their products, such as lipopolysaccharide (LPS), not only to enter the sterile submucosa and activate immune cells through the binding and recognition of their PAMPs in the respective PRRs but also favour microbial translocation, via current blood, to other organs and tissues, characterizing extraintestinal manifestations (EIM), with an impact on the functional status of the patients and their quality of life (Fig. 2) (Knutson et al., 2013; Hussein et al., 2008).
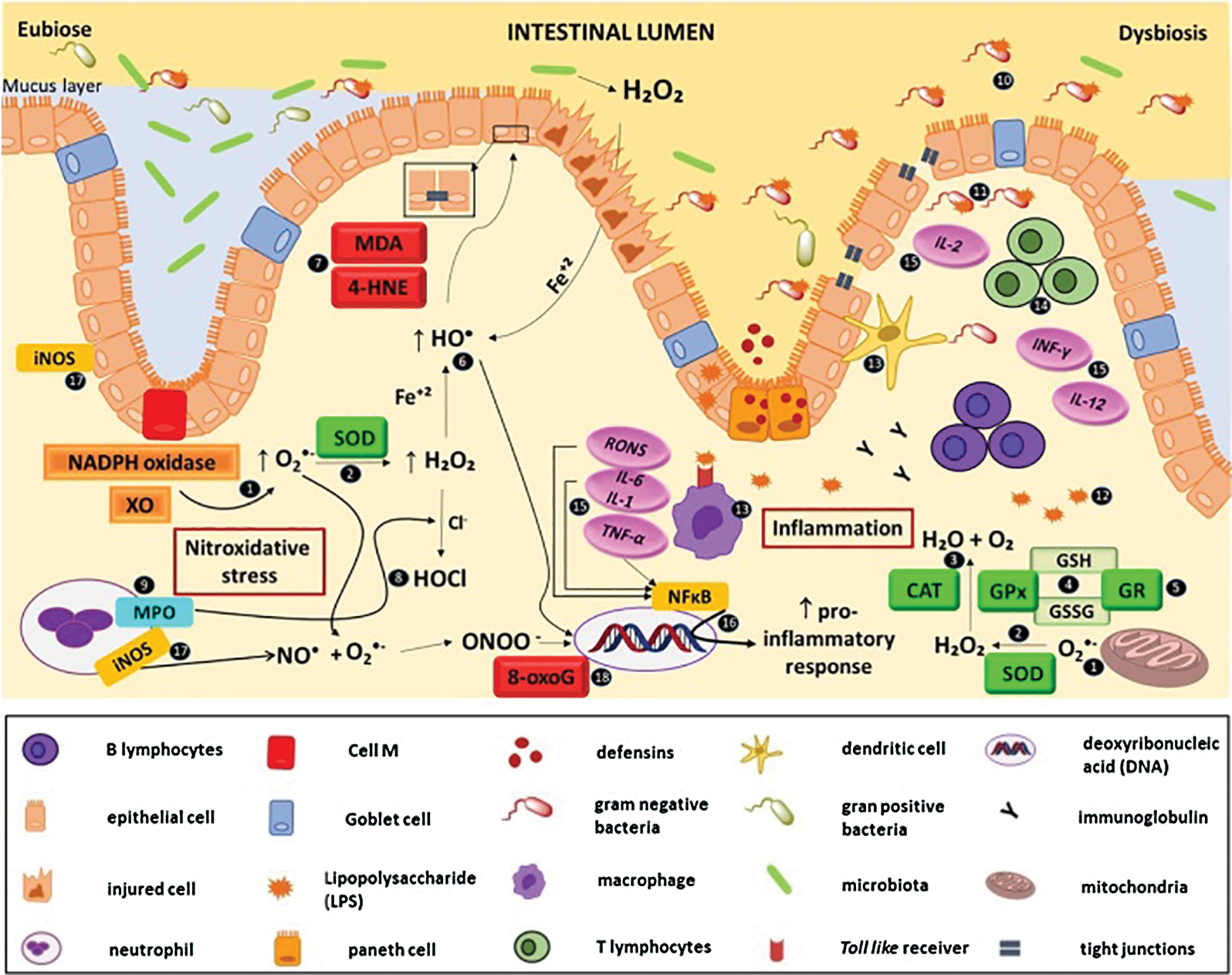
Figure 1: Interaction between dysbiosis, nitroxidative stress and inflammation in inflammatory bowel disease.
EIM can be identified in 25–40% of patients with IBD. The variation between remission and activity of symptoms is linked to morbidity and mortality. The main described manifestations include those that affect the liver and bile ducts, the skin, joints, eyes, and blood vessels. The heart, brain, and kidney are also affected (Annese, 2019; Garber and Regueiro, 2019). In addition, a common link regarding the microbial composition/nitroxidative stress of IBD and some diseases involving extraintestinal organs has been reported. This review aims to elucidate the participation of nitroxidative stress and dysbiosis in EIM genesis.
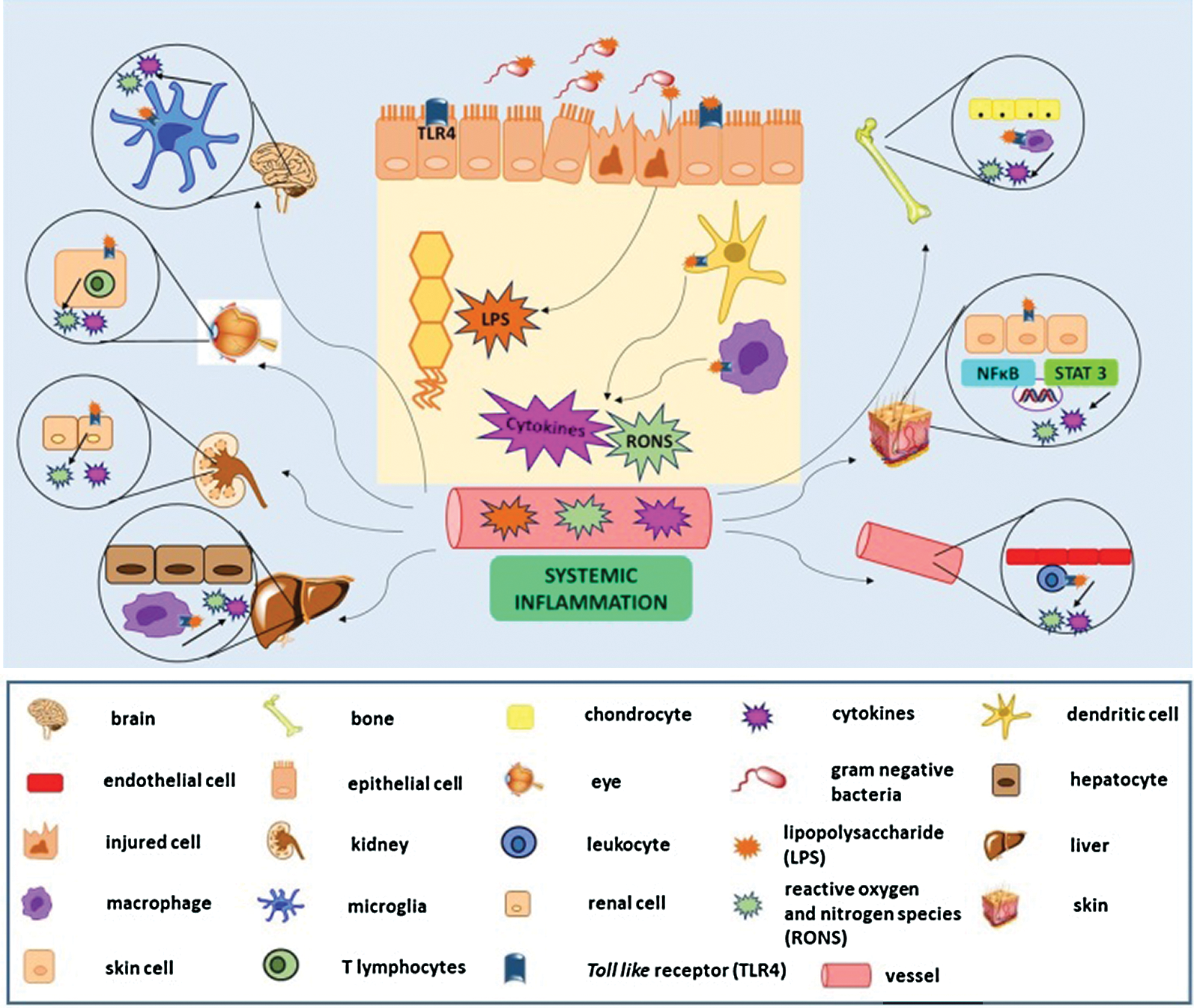
Figure 2: Microbiota translocation in inflammatory bowel disease promotes systematic inflammation and affects several tissues, causing local production of reactive oxygen and nitrogen species (RONS) and cytokines.
A narrative review of the literature was carried out on the association of inflammation, oxidative and nitrosative stress, and dysbiosis in the onset of EIM of IBD using the PubMed database. Systematic reviews, in vivo and in vitro studies (rats and mice) were included, with a total of 200 papers. Studies carried out on dogs, rabbits, pigs, or monkeys were not included in this review. The following keywords were used: oxidative stress, intestinal microbiota, dysbiosis, ulcerative colitis, Crohn’s disease, inflammatory bowel disease, liver, hepatic, joint, skin, eyes, ophthalmological, thromboembolism, cardiovascular diseases, heart, brain, cerebral, neurologic, renal, kidney, lipopolysaccharide, endotoxin.
After an extensive review, the results are displayed in the following topics: hepatobiliary; osteoarticular, dermatological and ophthalmological; thromboembolic; cardiovascular; neurological; and renal manifestations.
Hepatobiliary manifestations (HM) comprise one of the most common repercussions in IBD, with a prevalence of 3–50%, especially in those with UC. In addition, up to 5% of adults with IBD will develop some liver disease (Mendes et al., 2007; Restellini et al., 2017; Silva et al., 2019; Venkatesh et al., 2011). Primary sclerosing cholangitis (PSC) and non-alcoholic fatty liver disease (NAFLD) are the most common forms and can occur at any time during the course of intestinal disease or before its diagnosis (Annese, 2019; Fousekis et al., 2018).
PSC is a progressive chronic cholestatic disease, characterized by inflammation, stenosis, fibrosis, and obstruction of the intra- and extrahepatic ducts, which can progress with complications such as cirrhosis, liver failure, and portal hypertension, in addition to an increased risk of cholangiocarcinoma and colorectal cancer (Eaton and Talwalkar, 2013; Maggs and Chapman, 2008; Tsaitas et al., 2014). Epidemiological studies demonstrate that 60–80% of patients with PSC have IBD, especially UC (approximately 75%), and up to 8% of patients with IBD have PSC (Hirschfield et al., 2013; Mendes et al., 2007; Tsaitas et al., 2014).
NAFLD, in turn, characterized by excess fat deposited in the liver, is responsible for up to 40% of the hepatic repercussions of the non-alcoholic subtype in patients with IBD (Magri et al., 2019; Vernon et al., 2011). The prevalence is higher when compared to the general population, ranging between 1.5% and 55% (Chao et al., 2016; Palumbo et al., 2019).
The link between HM and IBD is still not well understood; however, multiple factors are involved, including genetic predisposition (HLA-B8, HLA-DRB1* 0301, HLA-DRB3* 0101, HLA-DRB1* 0401, REL, IL2 and CARD9) and hepatotoxicity induced by the use of drugs (corticosteroids, aminosalicylates, methotrexate, thiopurine, and antitumour necrosis factor-alpha) (Chapman et al., 2010; Eaton and Talwalkar, 2013; Janse et al., 2011; Karlsen et al., 2010). However, the role of intestinal dysbiosis, inflammation, and nitroxidative stress also stands out, resulting from immune-mediated processes (Fousekis et al., 2018; Navaneethan, 2014; Restellini et al., 2017).
Bacterial translocation, evidenced by the significant increase in the levels of endotoxins and LPS in the portal vein in humans and animal models, implies the participation of the intestinal microbiota in the pathogenesis of HM (Nakamoto et al., 2019).
Changes in the intestinal microbiota and dysfunction of the epithelial barrier play a crucial role in PSCs concomitant with IBD, as evidenced in humans. Intestinal dysbiosis–generally called dysbiosis– is an inherent characteristic of IBD, participating in the perpetuation and maintenance of chronic intestinal inflammation (Fujimoto et al., 2013; Nishida et al., 2018).
In humans, cohort studies have reported changes in microbial diversity and composition and have identified distinct phenotypes among patients with PSC and/or IBD when compared to healthy controls. It is noteworthy the significant decrease in the strain Faecalibacterium prausnitzi, present in both PCS and patients with IBD (Bajer et al., 2017; Quevrain et al., 2016; Sabino et al., 2016). This dysbiosis may be associated with an unregulated mucosal immune response and altered permeability that directs a local and extraintestinal inflammatory response through bacterial translocation. It has been suggested that in the liver, endotoxins, especially LPS, bind to Toll-like receptor 4 (TLR 4) and activate dendritic cells and macrophages, which are involved in the secretion of pro-inflammatory cytokines, such as tumour necrosis factor-alpha (TNF-α) and reactive oxygen and nitrogen species (RONS); the expression of adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) and vascular adhesion molecule 1 (VCAM-1); and in the initiation and progression of the fibrotic process characteristic of PSC (Reyes-Gordillo et al., 2017).
In an animal model of PSC, dysbiosis was associated with changes in the enterohepatic circulation of bile acids, damage to the mucus layer, reduced expression of tight junction proteins, and increased bacterial translocation (Liao et al., 2019).
Furthermore, a review showed that activation of the inflammasome nucleotide-binding domain, leucine-rich containing family, pyrin domain-containing-3 (NLRP3) in the intestine-liver axis by bacterial products contributed to the progression of liver damage through secretion of interleukin (IL)-1β and IL-18 via caspase-1 activity (Liao et al., 2019), which is related to the amplification of hepatocellular damage (Zmora et al., 2017).
Intestinal dysbiosis has also been recognized as a predisposing factor for NAFLD. The binding of bacterial DNA to TLR9 in Kupffer cells has been reported in animal studies to stimulate the secretion of IL-1β, which amplifies steatosis and liver fibrosis, in a MyD88-dependent mechanism (Miura et al., 2010; Muruve et al., 2008; Purchiaroni et al., 2013).
The involvement of Klebsiella pneumoniae in NAFLD, a gram-negative bacterium associated with lung infections, has also been investigated. Recurrent infections by this resistant pathogenic bacterium stimulate an influx of pro-inflammatory cytokines in the colon and ileum, and the increased colonization of K. pneumoniae induced UC in an animal model, where it was able to increase the expression of cyclooxygenase- 2 (COX-2), IL-6, IL-1β and TNF-α and the levels of nitric oxide (NO•) and reduce tight junction proteins (Kaur et al., 2018; Lee and Kim, 2011; Zhou et al., 2009). Additionally, a study in mice demonstrated that these animals developed liver steatosis after faecal microbiota transplant containing K. pneumoniae isolated from a patient with NAFLD. Lastly, a cohort study reported a strong association between this bacterium and the severity of NAFLD in 61% of individuals. This result suggested that dysbiosis causes this condition due to excessive alcohol production, as this bacterium is associated with ethanol production in the faeces of patients with NAFLD when compared to healthy patients, and, consequently, ROS production and mitochondrial dysfunction (Chen et al., 2020; Yuan et al., 2019).
In dysbiosis, redox imbalance plays a crucial role in the damage, apoptosis/necrosis, and formation of toxic aldehydes (lipid peroxidation) (Leung and Nieto, 2013; Shearn et al., 2018).
Significant periportal inflammation in the PSC marked by neutrophilic infiltrate and activation of NADPH oxidase is related to the generation of elevated levels of lipid peroxidation products and derivatives, such as malondialdehyde (MDA), acrolein, and 4-hydroxy-2-nonenal (4 HNE), which are important markers in the liver associated with protein and deoxyribonucleic acid (DNA) damage. Toxic aldehydes can cause post-translational changes in certain proteins, causing protein carbonylation, which determines the toxicity and degree of inflammation/fibrosis in the liver (Osna et al., 2016; Shearn et al., 2018; Shearn et al., 2019).
In an animal model of PSC, an increase in periportal oxidative stress depending on the stage of cholestasis was demonstrated (Shearn et al., 2019). Corroborating these findings, human studies carried out in patients with PSC concomitant with IBD showed an increase in periportal oxidative stress, with the elevated presence of toxic aldehydes, inflammation (↑ lymphocytes, ↑ Kupffer cells and ↑ MPO), and unregulated antioxidant response (Shearn et al., 2018).
Additionally, the increase in intestinal permeability observed in IBD patients allows lymphocytes, activated in the intestine, to enter the enterohepatic circulation and cause inflammation in the liver. This promotes the recruitment of adhesion molecules and chemokines, such as MAdCAM-1 (mucosal vascular addressin cell adhesion molecule-1) and CCL25 (C-C motif chemokine ligand 25), which are chemotactic for other inflammatory cells, such as macrophages and dendritic cells (Adams and Eksteen, 2006; Eksteen et al., 2004; Grant et al., 2002).
In NAFLD, increased β-oxidation in peroxisomes and microsomes generates hydrogen peroxide (H2O2) not coupled to phosphorylation and cytochromes P4502E1 and P4504A, respectively (Bellanti et al., 2017; Robertson et al., 2001). The imminent redox imbalance and the elevation of lipids, associated with glutathione reductase (GR) depletion, can lead to the formation of intermediate lipids and cause stress of the endoplasmic reticulum, which in turn are related to inflammation and apoptosis (Bellanti et al., 2017; Higa and Chevet, 2012; Mari et al., 2006; Pagliassotti, 2012).
In humans, it was seen that the accumulation of lipids in the liver, especially cholesterol, alters the cellular redox status by prominently activating the oxidative pathways in the mitochondria, causing a significant increase in the formation of ROS that surpasses the neutralizing capacity of endogenous antioxidant defenses, leading to mitochondrial dysfunction (Muriel, 2009; Serviddio et al., 2011; Sunny et al., 2011).
Moreover, lipid peroxidation products, due to overproduction of ROS, influence the progression of NAFLD by complex inflammatory mechanisms, including inhibition of the peroxisome proliferator-activated receptors (PPARs) and TLR 7 pathways and activation of nuclear factor kappa B (NFκB) and activating protein 1 (AP-1) Green and Wahli (1994). These factors stimulate the production of pro-inflammatory cytokines, especially TNF-α and IL-1β, which increase the expression of sterol regulatory element-binding protein (SREBP), responsible for the transcription of genes that encode enzymes involved in the synthesis of lipids and the later appearance of steatosis (Bellanti et al., 2017; Chen et al., 2008; Kohjima et al., 2007).
Therefore, the involvement of dysbiosis/inflammation/nitroxidative stress as mediators of hepatobiliary lesions triggered by IBD seems to be increasingly consistent and should be considered in clinical practice.
Osteoarticular, dermatological and ophthalmological manifestations
These manifestations occur even before the diagnosis of UC, correlating or not with disease activity (Olpin et al., 2017). Genetic susceptibility has been proposed to explain the link between then and IBD, such as TNF-α and variations in TNF receptor-associated factor interacting protein 2 (TRAF3IP2) that occur in erythema nodosum (Ciccacci et al., 2013; Orchard et al., 2002; Suh et al., 2019; Timani and Mutasim, 2008). In this context, in some of these diseases, genetic alterations related to microbiota disruption have been described, including ankylosing spondylitis and uveitis, which were identified as the presence of B27 human leukocyte antigen (HLA-B27) that predisposes individuals to dysbiosis (Ciccia et al., 2016; Costello et al., 2015; Hermann et al., 1993; Lin et al., 2014a; Pimentel-Santos et al., 2013; Sheth et al., 2015; Speca and Dubuquoy, 2017).
However, genetic factors alone are unable to elucidate the pathophysiological pathways, suggesting the involvement of environmental factors. It has been proposed that intestinal lumen antigens can form circulating immune complexes and could be deposited in these organs, causing an inflammatory response and RONS production, which are responsible for the characteristic lesions of the disease (Lin et al., 2014a; Lin et al., 2014b; Yang et al., 2016).
Osteoarticular manifestations: Spondyloarthropathies (SpA), including ankylosing spondylitis and peripheral arthritis, are found in 10–39% of patients with IBD, especially in patients with CD (Fantini et al., 2009; Gionchetti et al., 2015; Karreman et al., 2017). Some mechanisms are described to explain this association. One of these findings suggests that lymphocytes and macrophages activated in the Payer plate and in the mesenteric lymph nodes start to express and stimulate cell and vascular adhesion molecules (α4β7 and αΕβ7 integrins, cadherin E, vascular adhesion protein 1 [VAP-1], ICAM-1) that promote the adherence of these immune cells in the synovial endothelium and inside the joints, releasing cytokines directed to synovial fibroblasts. These findings were corroborated in samples from the affected part of the intestine of patients with CD and CU, where it was demonstrated that the leukocyte populations of the inflamed intestine were avidly bound to synovial vessels, evidenced by the expression of adhesion molecules and ligands (ICAM-1 and P-selectin) (Jacques and Elewaut, 2008; Salmi and Jalkanen, 2001).
Another link described in some studies suggests that the mechanisms of IBD-associated SpA involve a hypothesis of “intestine-synovia-joint axis”, including changes in the intestinal microbiota and activation of T cells in the intestine for liquid synovial joint in genetically predisposed individuals (Arvikar and Fisher, 2011; Fragoulis et al., 2019; Brakenhoff et al., 2010).
In this context, germ-free and transgenic animals for the HLA-B27 gene (related to joint disease) did not develop inflammation or lesions characteristic of colitis and spondyloarthritis (Gilis et al., 2018; Scher et al., 2016; Taurog et al., 1994).
Additionally, it has been reported in patients with IBD and SPA overexpression of E-cadherin, the major component of adhering junctions, responsible for maintaining intestinal homeostasis and barrier function, indicating the participation of microbiota/endotoxins in these conditions (Demetter et al., 2000; Demetter et al., 2005).
These changes in microbiota and intestinal permeability allow the translocation of bacteria and endotoxins, such as LPS, to the synovial fluid and joints, causing local inflammation (Asquith et al., 2014). Dysbiosis induces the M1 phenotype in macrophages, which are responsible for increasing the production of pro-inflammatory cytokines (Yang et al., 2016). Ciccia et al. indicated in intestinal biopsy samples from patients with SpA that infiltrating monocytes were responsible for the increased IL-23 expression, and overexpression of IL-23 is an important characteristic of subclinical gut inflammation in SpA. Further, to confirm this theory of “intestine-synovia-joint axis”, a reduction in the number of Faecalibacterium praustnizii species, which are beneficial species for intestinal health and the immune system, has been described in the faeces of patients with SpA and IBD (Tito et al., 2017).
Together, these findings (in experimental and human studies) suggest the involvement of microbiota homeostasis in the immune response and molecular mimicry in patients with SpA and IBD.
Osteoporosis is another osteoarticular disease common in patients with IBD and has been identified in 18–42% of these, increasing the fracture risk, especially in older patients (>60 years). However, it is not clear if IBD patients have an increased risk of reduced bone mineral density (BMD) (van Hogezand and Hamdy, 2006).
Osteoporosis in IBD is multifactorial, but medications used in IBD treatment seems to be the main cause of BMD in these patients. Chronic steroid therapy may decrease BMD, damage bone tissue structure, increase the risk of fractures (van Staa et al., 2005), and, by the other side, decrease intestinal calcium absorption and calcium kidney reabsorption, leading to an increase in the parathormone level, responsible for the stimulation of osteoclasts and increasing bone loss (van Staa, 2006).
The link between osteoporosis/IBD/oxidative stress/microbiota is not evident. However, in a recent review of Ratajczak et al. (Ratajczak et al., 2020), the authors did an association between vitamin C deficiency and risk of osteoporosis in IBD patients. According to then, as an antioxidant, vitamin C: (1) Decreases the level of ROS, which increase bone resorption, throughout the activation of (NF-κB) which is a crucial mediator of TNF-α and osteoclastogenesis; (2) Creates a redox state and modulates gut microbiota increasing Firmicutes and a decreased Bacteroides level. All dates together suggest an important role of antioxidant status in bone health to IBD patients. However, the exact mechanism is unclear and must be studied posteriorly.
Dermatological manifestations: Erythema nodosum and pyoderma gangrenosum are the most frequent EIM on the skin of IBD (Greuter and Vavricka, 2019; Vavricka et al., 2011). Nodosum erythema is the most common symptom, more present in CD than in UC, especially in women (Farhi et al., 2008; Vavricka et al., 2011; Vavricka et al., 2015). Its relationship with IBD is more associated with genetic factors (Suh et al., 2019; Timani and Mutasim, 2008).
Gangrenous pyoderma, in turn, mainly affects patients with UC to the detriment of those with CD and can appear before or after the diagnosis of IBD (Ahn et al., 2018; Annese, 2019). The pathophysiological link is still unclear, but an unregulated inflammatory response is proposed, involving neutrophils and T cells against antigens common to the organs. Data from immunohistochemistry and analysis of proteins in necrotic tissue in patients with gangrenous pyoderma have demonstrated overexpression of the TNF-α, NF-κB, and signal transducer and activator of transcription 3 (STAT3) pathways (Vavricka et al., 2015).
Psoriasis, a chronic inflammatory skin disease, has also been associated with IBD and is more prevalent in patients with CD than in those with UC (Fu et al., 2018; Suh et al., 2019). A recent meta-analysis also showed that psoriasis was significantly related to IBD (Fu et al., 2018), being more prevalent in these patients than in the general population.
Despite the genetic link (IL23R and IL12B), it has been proposed that the intestinal microbiota plays an important role in this context (Capon et al., 2007; Cho, 2008; Cottone et al., 2019; Ellinghaus et al., 2012). The decrease in the genus Faecali bacterium prausnitzii (a beneficial bacterium for the maintenance of intestinal homeostasis, as already mentioned in this review), observed in both patients with CD and psoriasis, demonstrates the relevance of an interaction between the microbiome and an adequate immune response (Eppinga et al., 2016).
Ophthalmological manifestations: Ophthalmological manifestations may be present in 4–12% of patients with IBD; however, the reported prevalence rate can reach up to 29%, especially in CD. The most common conditions reported are anterior uveitis and episcleritis, present in 0.3–10% of patients with IBD (Greuter and Vavricka, 2019; Harbord et al., 2016; Larsen et al., 2010; Taleban et al., 2016).
The hypothesis of the microbiota-intestine-eye has been proposed and investigated. In animal models of autoimmune uveitis, it has been shown that dysbiosis induces the activation of retinal-specific autoreactive T cells and intraocular inflammation (Horai and Caspi, 2019; Horai et al., 2015; Nakamura et al., 2016). In addition, there was an increase in intestinal permeability and antimicrobial peptide expression concomitant with the effector T cell response in the initial stage of uveitis (Janowitz et al., 2019).
In addition, nitroxidative stress, identified through MDA elevation levels in the aqueous humour and decreased antioxidant defense (superoxide dismutase [SOD], catalase [CAT] and glutathione peroxidase [GPx]), has been confirmed in an endotoxin-induced anterior uveitis model (Rahman and Biswas, 2004).
Recent cohorts, including individuals with autoimmune anterior uveitis, identified changes in the composition of the intestinal microbiota and a faecal metabolic phenotype that significantly differed when compared to that of healthy controls (Huang et al., 2018; Chakravarthy et al., 2018). However, the scarcity of studies in humans that have evaluated the intestine in eye diseases hinders the effective association.
Despite these molecular findings suggesting the close link between microbiota, nitroxidative stress, intestinal inflammation and osteoarticular, dermatological and ophthalmological manifestations in patients with IBD, these crosslinks are not yet well understood, and additional studies, especially in humans, are still needed.
The prothrombotic state is considered a characteristic of patients with IBD, especially during the symptomatic phase (Nguyen et al., 2014). The most common manifestations are deep venous thromboembolism and pulmonary embolism (van Assche et al., 2013). Chronic inflammation (monocyte chemoattractant protein-1 [MCP-1], IL-6 and IL-8) activates hemostasis and causes hypercoagulation and, consequently, abnormalities in the microvascular tissue characteristic of endothelial dysfunction (Danese et al., 2007; Esmon, 2005; Levi et al., 2012; Papa et al., 2008; Roifman et al., 2009; Zezos et al., 2014).
Several factors cause lesions in the gut microvascular endothelium, among which the most important described are the bacterial endotoxins present in the gut lumen, pro-inflammatory cytokines, and hypoxia (Danese, 2007; Joseph et al., 2002; Levi et al., 2012; Makrides, 1998; Stadnicki, 2012; Zezos et al., 2014).
Once activated, the vascular endothelium starts to express a large amount of cell adhesion molecules (ICAM-1, VCAM-1, and PCAM-1) and adhesins (selectins and integrins) that allow the recruitment and transmigration of leukocytes through the vessel, platelet activation, and aggregation (Davis et al., 2003; Panes and Granger, 1998; Schuermann et al., 1993). This scenario promotes the coagulation cascade, with an evident increase in the factors involved in this process (factors V, VIII, von Willebrand, and fibrinogen), thus enabling the formation of thrombi. Vascular and tissue damage spread through a vicious cycle characterized by an increase in the production of cytokines, especially TNF-α, IL-6 and vascular endothelial growth factor (VEGF), RONS and chemokines (Danese, 2007; Hudson et al., 1992; Jorens et al., 1990; Scaldaferri et al., 2011; Zezos et al., 2014). Increased expression of VEGF and its receptor was seen in samples from patients with IBD when compared to healthy individuals (Scaldaferri et al., 2009).
Furthermore, vascular dysfunction in IBD was associated with an imbalance in RONS levels. Inflammatory and immunological stimuli, bacteria and LPS have been shown to activate inducible nitric oxide synthase (iNOS), which induces the production of large amounts of •NO–a potent vasodilator and anti-aggregation agent that causes indirect harmful effects through the generation of other species, such as nitroxyl anion (NO−) and peroxynitrite (−OONO), which is able to cause lipoperoxidation and damage to DNA molecules (Beckman and Koppenol, 1996; Kolios et al., 2004). Studies with colonic biopsies of patients with CD and UC, showed overexpression of iNOS in the inflamed epithelium and nitrotyrosine, suggesting that this finding is associated with the formation of −OONO and the nitration of cellular proteins (Singer et al., 1996; Dijkstra et al., 1998).
In addition, literature reviews show that in the endothelial cells of the chronically inflamed microvasculature, there is a decrease in the production of NO•, mainly due to the selective inhibition of iNOS and the non-selective inhibition of the constitutive forms of nitric oxide synthase (NOS), and an increase in superoxide anion radical (O2•-), which was proportional to the increase in recruitment and leukocyte adhesion (Binion et al., 1998; Binion et al., 2000; Hatoum et al., 2003).
The increase in the serum levels of homocysteine (HCy), a sulfur-containing amino acid resulting from the demethylation of methionine, is also associated with an elevated risk of venous thromboembolism by inducing platelet activation in the endothelium and increasing prothrombotic components (Zhang et al., 2014). In a dysbiotic environment, the bacteria that synthesize methionine contribute to excessive HCy formation (Kurilshikov et al., 2019).
Intestinal dysbiosis and bacterial translocation have also been linked to endothelial dysfunction. A review described that, in the bloodstream, microbial endotoxins present in the outer membrane of gram-negative bacteria, especially LPS, bind to TLR4 in immune cells, forming a complex that binds to the MD-2 protein and CD14. This complex formation results in the production of pro-inflammatory cytokines and other mediators involved in endothelial damage, the procoagulant state, the recruitment and transformation of macrophages into foam cells, and the initiation of atherosclerotic plaque (Szeto et al., 2018a; Szeto et al., 2018b).
Therefore, the activation of the endothelium associated with chronic bowel inflammation and dysbiosis may be critically implicated in triggering hypercoagulability and subsequent onset of thromboembolic events.
Patients with IBD have a high risk of coronary artery disease, in part, attributed to a greater susceptibility to the occurrence of thromboembolic events and nutrient malabsorption, especially selenium (Benstoem et al., 2015; Castro Aguilar-Tablada et al., 2016; Hansson, 2005; Wu et al., 2017). On the other hand, it has been suggested that the risk of atherosclerosis and other cardiovascular diseases is associated with endothelial dysfunction caused by systemic inflammation, dysbiosis, and nitroxidative stress (Aniwan et al., 2018; Bigeh et al., 2019; Horowitz et al., 2007; Wu et al., 2017).
Systemic inflammation in IBD leads to RONS generation and nitroxidative stress (Wu et al., 2017; Zanoli et al., 2015). A study in patients with CD observed high levels of pro-inflammatory molecules such as TNF-α, IL-1, IL-6 and C-reactive protein (CRP) contribute to endothelial dysfunction, characterized by vascular smooth muscle cell hyperplasia and decreased •NO production, resulting in a reduction in vessel relaxation. This induces the infiltration of neutrophils into the blood vessels, causing changes in the smooth muscle cell phenotype, an increase in matrix metalloproteinase production, and a decrease in elastin and collagen fibres due to the activation of collagenases and elastases, thus leading to the formation of rigid fragments that induce atherosclerotic processes (Schinzari et al., 2008). Moreover, TNF-α signaling pathways induce the expression of osteoblast markers (osteocalcin and osteopontin) in endothelial cells, which leads to an increase in calcification and reduced vascular elasticity, resulting in arterial stiffening, coronary artery disease, and heart failure (Floege and Ketteler, 2004; Wu et al., 2017; Zanoli et al., 2015; Zieman et al., 2005).
Intestinal dysbiosis, in turn, associated with increased permeability, allows bacterial translocation. Has been demonstrated in in vitro assays (Howell et al., 2011; Maziere et al., 1999) and an animal model (Wiesner et al., 2010) that high levels of LPS and bacterial products directly induce the atherosclerotic process through the formation of oxidized low-density lipoprotein (oxLDL), a key atherosclerotic lesion component, activation of macrophages and adhesion molecules and, consequently, the formation of foam cells and stimulation of pro-inflammatory cytokine expression. The latter, in association with other cellular mediators such as VEGF, CRP, and platelets, cause endothelial dysfunction and eventual atherosclerosis.
A recent cohort study exploring the relationship of intestinal microbiota with plasma metabolites, cardiovascular metabolic risk score, and cardiometabolic phenotypes demonstrated that L-methionine-producing bacteria were associated with atherosclerosis in obese individuals. It is suggested that methionine causes this effect through its direct conversion to HCy, contributing to serum elevation, which is a known cardiovascular risk factor that will be addressed later (Kurilshikov et al., 2019).
The presence of bacterial DNA in human plasma has been addressed in some reviews and associated with endotoxin levels to trigger systemic inflammation and increase the instability of atherosclerotic plaques. It binds to TLR-9, stimulating inflammatory intracellular signaling pathways such as mitogen-activated protein kinase (MAPK), NF-κB, PI3-kinase, and Jun N-terminal kinase (El Kebir et al., 2008; Szeto et al., 2018a).
The intestinal microbiota is also related to the trimethylamine/N-trimethylamide (TMA/TMAO) pathway (Hansen et al., 2015; Wang and Zhao, 2018). Proteus mirabilis is a gram-negative bacterium, a component of the fecal microbiota, but in a dysbiotic environment, it is found in higher numbers. These species, from the carbon extraction of some compounds such as choline, phosphatidylcholine, glycerol phosphocholine, carnitine, betaine, and γ-butyrobetaine, produce the metabolite TMA by TMA lyases (Tang and Hazen, 2014; Wang et al., 2015; Wang and Zhao, 2018). TMA is oxidized to TMAO in the liver by flavin monooxygenase and transported to the systemic circulation (Bennett et al., 2013; Koeth et al., 2013; Tang and Hazen, 2014; Wang and Zhao, 2018) and activates smooth muscle cells of the vessels, endothelial MAPK and the NF-κB pathway, leading to the expression of pro-inflammatory cytokines and leukocyte adhesion in addition to inducing transformation of macrophages to foam cells to activate NOD (nucleotide-binding oligomerization domain), LRR (leucine-rich repeats) and NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) (Chen et al., 2017; Seldin et al., 2016; Wang and Zhao, 2018; Wei et al., 2017). Additionally, TMAO reduces the expression of the genes CYP7A1 (cytochrome P450 family 7 subfamily A member 1) and CYP27A1 (cytochrome P450 family 27 subfamily A member 1), affecting the metabolism of cholesterol and bile salts and the release of calcium in the platelet endoplasmic reticulum, facilitating the formation of thrombi (Koeth et al., 2013; Zhu et al., 2016).
Furthermore, hyperhomocysteinaemia is an independent cardiovascular risk factor and has been observed in both patients with UC and CD at concentrations 4–5 times higher than those in healthy individuals (Drzewoski et al., 2006; Oussalah et al., 2011; Tyagi et al., 2005; Wu et al., 2017).
High levels of HCy cause nitroxidative stress and endothelial dysfunction through self-oxidation catalysed by cationic metals, which results in the formation of O2•- and, when reacting with •NO, forms (ONOO-), which reduces the bioavailability of •NO (Sen et al., 2010; Tyagi et al., 2005). Studies in cell culture (Liu et al., 2013; Zhu et al., 2016) and animals (Sen et al., 2010) have shown that high levels of HCy induce RONS formation, causing an increase in the concentrations of H2O2 and MDA and mitochondrial damage by decreasing the expression of cytochrome c oxidase III/ATPase 6/8 and heat shock protein 60 and impairing the antioxidant defence system (GPx, hemeoxygenase and SOD), thus leading to redox imbalance (Liu et al., 2013; Tyagi et al., 2005). It also activates the NFκB pathway, adhesion molecules (ICAM-1, VCAM-1) and MCP-1. These, in turn, interact with inflammatory cells of the endothelium, leading to the atherosclerotic process (Silverman et al., 2002; Wang et al., 2002).
Malabsorption, nutritional deficiencies (vitamins B1, B12, D and E, folic acid and nicotinamide), infections, thromboembolism, adverse drug effects (metronidazole, sulfasalazine, steroids, cyclosporine), and immunological abnormalities are related to changes in the bowel-brain axis (Casella et al., 2014; Moris, 2014). The pathophysiology of neurological manifestations of IBD and their prevalence are not yet fully understood. Neurologic and neuromuscular complications in IBD (peripheral neuropathies, multiple sclerosis, cerebrovascular diseases, and others including psychiatric disorders) have been estimated from 0.25 to 35.7% due to the different forms of diagnosis (Elsehety and Bertorini, 1997; Gondim et al., 2005; Lossos et al., 1995).
A link has been proposed between chronic intestinal inflammation and neuropathy onset. The deregulated immune response involving mainly T cells towards the autoantigens that crossed the blood-brain barrier (BBB) induces central nervous system activation of astrocytes and microglia, which stimulates the production of pro-inflammatory cytokines, RONS, glutamatergic excitotoxicity, and autoantibody generation against the myelin sheath, causing axonal injury and neuronal dysfunction (Casella et al., 2014; Moris, 2014; Singh et al., 2013). According to Nemati et al. (2019), the development of neurological disease in IBD may be coincidental (chance association) or the consequence of the primary disease and this differentiation is necessary because they may require different treatments. Furthermore, investigations in this context are not yet routine in clinical practice.
An intimate link between the microbiota-gut-brain axis has been proposed. The microbiota influences several brain activities, including the modulation of neuroimmune responses and maintenance of the integrity of the epithelial barrier and the BBB, through the expression of occludin and claudin, demonstrated in an animal model (Braniste et al., 2014). Changes in the microbiota and bowel permeability allow the spread of bacteria, toxins such as LPS, and other metabolites (TMAO, for example) towards the BBB. This causes an unregulated immune response, with activation of the microglia that alters its secretory profile and stimulates the production of pro-inflammatory chemokines and cytokines (MCP-1, interferon-gamma [IFN-γ], IL-6, IL-8, and TNF-α), as well as increases RONS (Block et al., 2007; Del Rio et al., 2017; Shemer et al., 2015).
Multiple sclerosis (MS), characterized by demyelination and neuroinflammation, has been closely related to IBD. Studies indicate that first-degree relatives of patients with MS are at risk for CD development (risk of 1.4), while IBD patients have a risk of 1.7 of developing MS (Gupta et al., 2005; Nielsen et al., 2008; Noseworthy et al., 2000). Changes in the microbiota in patients with MS have been identified in this process, such as an increase in antibodies against intestinal microbial components (Banati et al., 2013). Furthermore, in cell culture, the activation of mucosal-associated invariant T cells (MAIT), seen in IBD, also plays a role in MS, as they can interact with microbial components and stimulate the innate and adaptive response, including CD8+ T cells and the production of pro-inflammatory cytokines, such as IL-17. Moreover, these cells express high levels of CD161 (cluster of differentiation 161), CCR6 (chemokine receptor 6), and IL-18 (Moreira et al., 2017).
Neurodegenerative diseases, such as Parkinson’s disease (PD), have also been associated with intestinal dysbiosis and increased intestinal permeability, seen in IBD Animal studies have shown that the increase in LPS in UC causes an overactivation of microglia, TNF-α secretion and iNOS activation. The identification of α-synuclein (Parkinson’s disease–PD–marker protein) in the enteric nervous system and later in the neurons of the central nervous system (CNS) reinforced the link (de La Serre et al., 2015; Hoban et al., 2013; Qin et al., 2013; Villaran et al., 2010).
In humans, this relationship has also been confirmed. According to Villumsen et al. (2019), patients with IBD have up to 22% risk of developing PD when compared to healthy individuals.
The reduced integrity of the BBB in model animals and PD individuals has been reported, and it is possible that concomitant epithelial barrier dysfunction increases microbiota communication with CNS cells (Gray and Woulfe, 2015; Zhao et al., 2007).
On the other hand, studies have shown that patients with IBD have a 2–3 times higher risk of developing anxiety and depression than the general population, affecting 30% and 25%, respectively, of these patients, especially in the active phase (Mikocka-Walus et al., 2016; Walker et al., 2008).
The intimate connection and communication of the microbiota–gut–brain axis is proposed to explain this prevalence. The hypothalamic-pituitary-adrenal axis perceives and responds to the stimulus of stress and inflammation, leading to a decrease in the effector response of the vagus nerve and an increase in intestinal permeability. This favors bacterial translocation that activates the mucosal immune response, as well as other cells of the innate immune system in the brain, increasing microglia activity, pro-inflammatory cytokine levels and nitroxidative stress, which generate changes in brain functions and damage to neuroplasticity (Abautret-Daly et al., 2018; Bailey et al., 2006; Maes et al., 2012; Santos et al., 1999).
In cell culture and animal model assays, dysbiosis impairs its appropriate interaction with the host and influences neural activities in brain areas related to stress and behavior, with changes in brain-derived neurotrophic factor (BDNF) in the hypothalamus and amygdala, in addition to the decrease in SCFA-producing bacteria, which, in turn, are immunomodulatory metabolites. The participation of the microbiota seems to be related to the gamma-aminobutyric acid (GABA), serotonin and dopamine pathways, which are implicated in depressive and anxiety disorders (Bernstein, 2017; Foster and McVey Neufeld, 2013; Lyte et al., 2011).
The link between IBD and renal manifestations, although less frequent, has been demonstrated in both CD and UC. The reported diseases are nephrolithiasis, glomerulonephritis, and tubule-interstitial nephritis (Ambruzs et al., 2014; Corica and Romano, 2016).
Nephrolithiasis affects 12–28% of patients with IBD, and few studies report a 9–18% higher risk than in the general population. The frequency is higher in CD and patients submitted to surgical procedures such as colectomy with ileostomy and intestinal resection (Bianchi et al., 2018; Gkentzis et al., 2016; McConnell et al., 2002; Parks et al., 2003). In this context, a small cohort of 83 patients demonstrated an incidence of 24% IgA glomerulonephritis and 19% tubule-interstitial nephritis, especially in CD (Ambruzs et al., 2014).
Some links between renal manifestations and IBD have been described that seem to have an important role, including genetics by identifying the HLA-DR1 (human leukocyte antigen-DR1) and HLA-DR1/DQw5 genes present in both IgA glomerulonephritis and CD; adverse effects of drugs such as aminosalicylates and its derivatives, especially in tubule-interstitial nephritis; and a reduction in anti-lithogenic factors (citrate and magnesium) in nephrolithiasis, mainly due to chronic diarrhea (Ambruzs et al., 2014; Ganji-Arjenaki et al., 2017; Hueppelshaeuser et al., 2012; Kane, 2006; Oikonomou et al., 2011; Takemura et al., 2002; Worcester, 2002). However, these factors cannot fully explain the renal manifestations in patients with IBD.
In relation to nephrolithiasis, intestinal dysbiosis seems to be involved in the formation of calcium oxalate stones in the kidneys. It has been observed that individuals with IBD, who have a smaller population of Oxalobacter formigenes in their intestinal microbiota, have a high prevalence of nephrolithiasis (Kumar et al., 2004). O. formigenes is a gram-negative, anaerobic, commensal bacterium from the gastrointestinal tract that acts on the regulation of oxalate homeostasis; it degrades oxalate through the enzymes oxalyl-coenzyme A decarboxylase and formyl-coenzyme transferase and interacts with the intestinal mucosa, stimulating the secretion of endogenous oxalate into the lumen, a transport mechanism through the epithelium, thus contributing to the excretion of this compound (Arvans et al., 2017; Liu et al., 2017; Siener et al., 2013; Siva et al., 2009; Stewart et al., 2004).
It has been observed, especially in patients with CD, that decolonization of TGI by O. formigenes leads to a reduction in the intestinal catabolism of oxalate and the subsequent appearance of hyperoxaluria, the main mechanism involved in the formation of kidney stones. In addition, an increase in urinary oxalate was observed in these patients when compared to healthy controls. It occurs more frequently in individuals who have undergone ileum resection and colon-jejunal anastomoses (Hueppelshaeuser et al., 2012; Kane, 2006; Kumar et al., 2004; Oikonomou et al., 2011; Siva et al., 2009). The causes that lead to the decolonization of this bacterium have not yet been elucidated. However, it has been proposed that the possible reasons are precisely associated with changes in the composition of the intestinal microbiota, typical of IBD, which is the result of an inflammatory response exacerbated against these commensals, which results in a break in immunological tolerance (Kumar et al., 2004).
Nevertheless, other commensal bacteria, such as Lactobacillus and Bifidobacterium, also secrete enzymes capable of degrading oxalate in a medium that contains glucose and lactose (Campieri et al., 2001). The role of oxalate-degrading bacteria in the treatment of kidney stones has become of great interest in the scientific community, aiming at their use as a probiotic. Animal and human research using O. formigenes or their enzymes have shown promising results; however, data security is still required through well-controlled and larger-scale clinical trials. Other studies investigating the species Lactobacillus and Bifidobacterium have also shown a reduction in oxalate excretion, but the results are still controversial. The importance of additional standardized studies that determine the direct relationship between the decolonization of these bacteria and the presence of hyperoxaluria and consequent kidney stones and their influence as a risk factor for the identification of possible probiotics in the treatment of this condition is emphasized. It is said that functional and molecular approaches are needed to choose the best species that also have the ability to effectively colonize the intestine (Abratt and Reid, 2010; Mehta et al., 2016; Sadaf et al., 2017).
Oxalate generates toxic responses that result in the activation of phospholipase A2, which culminates in the production of arachidonic acid and various lysophospholipids. These factors, in turn, lead to mitochondrial dysfunction, an increase of RONS production, and the induction of changes in the expression of genes involved in the synthesis of molecules that inhibit the formation of calcium oxalate stones and in the activation of caspases, which are involved in apoptotic cell death (Cao et al., 2000; Cao et al., 2004; Jonassen et al., 2005; Oikonomou et al., 2011). In addition, the presence of TLRs, especially TLR4, expressed in renal epithelial cells and the identification of significant amounts of endotoxins in kidney stones suggest that this interaction generates activation of inflammatory pathways (Anders et al., 2004a; Anders et al., 2004b; McAleer et al., 2003; Oikonomou et al., 2011).
In IgA nephropathy, the intimate relationship of the microbiota-intestine-kidney axis has been implicated. The breakdown of the intestinal barrier and the increase in pro-inflammatory cytokines leads to bacterial translocation, facilitating the diffusion of microbial endotoxins and DNA through the bloodstream, which become established in the renal glomeruli. It was proposed that the deposited immune complexes react with luminal antigens and that there is a loss of exclusion and antigenic tolerance, sustained immune response and deregulation in the production and transport of IgA. Evidence shows that patients with CD present high levels of IgG and IgA due to an inflammatory response of the mucosa to K. pneumoniae (Anders et al., 2004a; Corica and Romano, 2016; Forshaw et al., 2005; O’Mahony et al., 1992; Takemura et al., 2002).
Finally, in tubule-interstitial nephritis, a deregulated immune and inflammatory response, the presence of autoantibodies and immunocomplexes against epitopes common to the intestine and kidney and molecular mimicry are likely pathophysiological mechanisms (Ambruzs et al., 2014; Corica and Romano, 2016; Fraser et al., 2001; Mahmud et al., 2002; Oikonomou et al., 2011; Poulou et al., 2006).
The diseases addressed may progress to renal failure and end-stage renal disease if the inflammation persists and/or is not contained. A few epidemiological studies have reported the incidence of renal failure between 2 and 15.9% in patients with IBD, and there is an elevated risk for end-stage renal disease in CD that is five times higher than that in CU and healthy controls, suggesting that IBD, especially CD, is an independent risk factor (Park et al., 2018; Primas et al., 2013). Some mechanisms have been proposed to explain the increased risk in patients with CD, including systemic inflammation resulting from the unregulated immune response in the intestine and the transmural character that can exacerbate renal damage, as has been reported with the increase in serum levels of IL-6 and CRP, in addition to autoimmunity (Cioffi et al., 2015; Fried et al., 2004). Therefore, it is essential to monitor renal function in patients with IBD to identify early signs of renal damage and guide the choice of appropriate and effective therapy.
There are few prospective studies in humans that assess the microbiota and nitroxidative stress in EIM of IBD. In some studies, associated comorbidities, such as diabetes mellitus and arterial hypertension, were not mentioned as a possible confounding factor. Thus, in this review, we propose a crosslink between redox imbalance and dysbiosis, using mainly in vitro and experimental reports, as well as physiological and biochemical established routes.
Prospects and future direction
Evidence suggests that chronic continuous nitroxidative stress and alteration of the intestinal microbiota may be strongly associated with the appearance of EIM inherent to patients with IBD (Fig. 3). Despite the involvement of genetic factors, it is necessary that other variables act to stimulate and/or to facilitate the expression of this phenotype, and the chronic inflammatory environment associated with increased intestinal permeability becomes decisive in determining these manifestations of the disease.
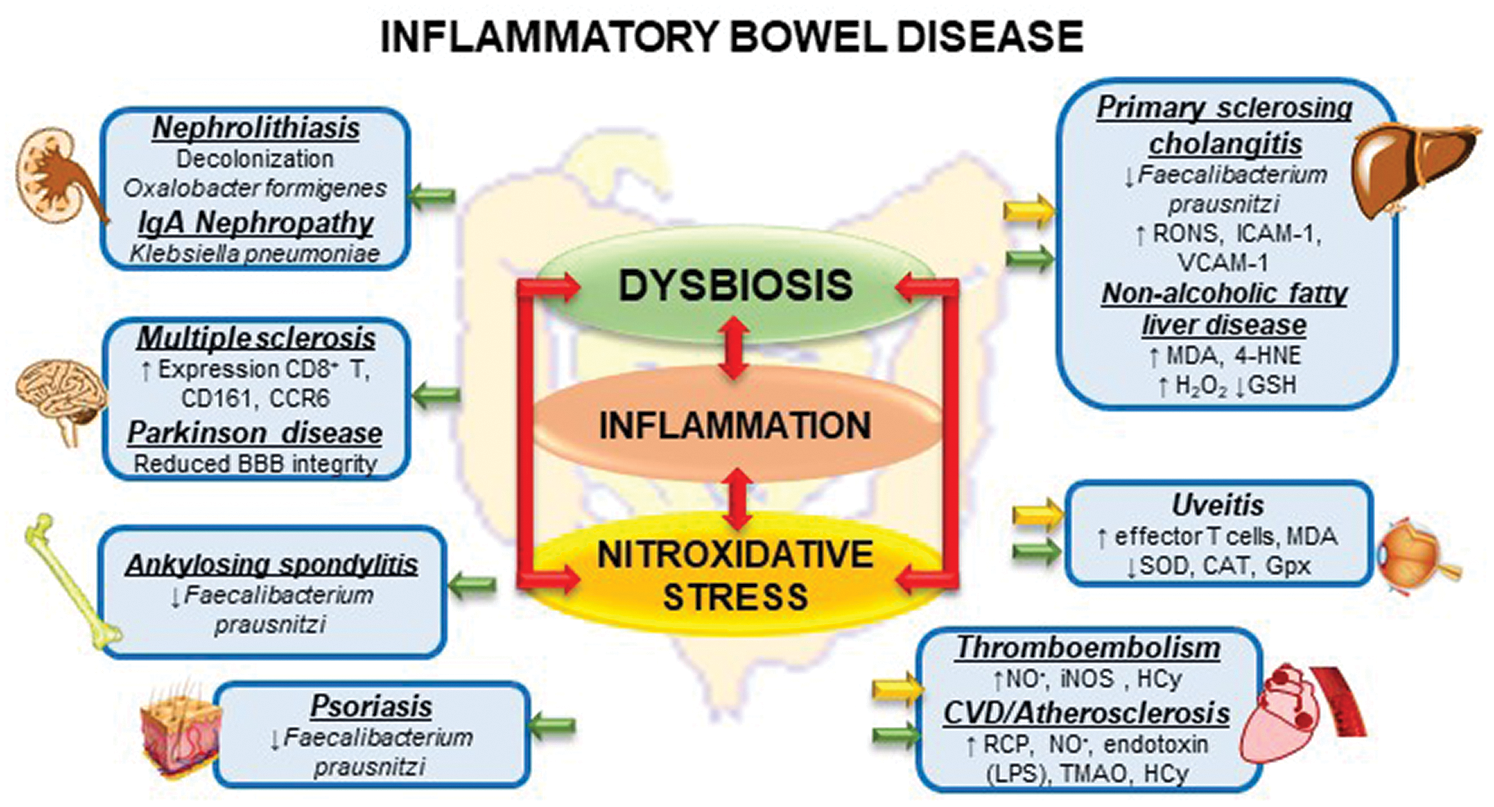
Figure 3: Interactions between dysbiosis, inflammation and nitroxidative stress and Extraintestinal Manifestation in inflammatory Bowel Disease.
However, the studies carried out in humans, herein described, demonstrate the association and the presence of dysbiosis in the hepatic (PSC), osteoarticular, and dermatological manifestations (psoriasis) and as a contributing factor for the formation of kidney stones. It is associated with the nitroxidative stress and lipid peroxidation in triggering NAFLD and endothelial dysfunction described in the pathophysiology of thromboembolic events. Nevertheless, according to the evidence presented, it is possible that an imbalance in the intestinal microbiota and nitroxidative stress are also implicated in cardiac, ophthalmological, and neurological outcomes, including psychological disorders. However, there are still gaps to explore in regard to the mechanisms and pathways that culminate in such conditions.
The absence of standardized clinical studies and the scarcity of large, prospective studies make it impossible to establish a cause-and-effect link, hindering a clinical intervention in this dimension. In view of this, well-designed clinical studies are necessary to further investigate the influence of nitroxidative stress and dysbiosis, as well as other factors mentioned in this work, such as the current pharmacological therapy used to trigger EIM. The understanding of the intrinsic pathophysiological pathways is urgently required for the development of effective preventive and therapeutic strategies aimed at manipulating the intestinal microbiota and controlling inflammation/nitroxidative stress.
Units: Units of measurement should be used concisely according to the International System of Units (SI). All units should be converted to SI units whenever possible.
Statistical Analysis: Appropriate statistical treatment of the data is essential. When statistical analysis is performed, the name of the statistical test used, the number for each analysis, the comparisons of interest, the alpha level and the actual p-value for each test should be provided.
Author Contribution: Amylly Sanuelly da Paz Martins (amyllymartins@gmail.com): Analysis and interpretation of data, writing, preparation of figures, and graphical abstract.
Samara Bomfim Gomes Campos (bomfim_samara@hotmail.com): Acquisition of data, library search.
Marília Oliveira Fonseca Goulart (mofg@qui.ufal.br): Critical evaluation of the content and contribution in conclusions and perspectives.
Fabiana Andrea Moura (fabianamoura_al@hotmail.com): Supervised all the steps, conception and design, figures and critical revision.
Funding Statement: This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq–Brazil) [435704/2018-4] and Fundação de Amparo à Pesquisa do Estado de Alagoas (FAPEAL)/PPSUS/Ministério da Saúde (MS) [60030-000876/2016].
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Abautret-Daly A, Dempsey E, Parra-Blanco A, Medina C, Harkin A. (2018). Gut–brain actions underlying comorbid anxiety and depression associated with inflammatory bowel disease. Acta Neuropsychiatrica 30: 275–296. DOI 10.1017/neu.2017.3. [Google Scholar] [CrossRef]
Abratt VR, Reid SJ. (2010). Oxalate-degrading bacteria of the human gut as probiotics in the management of kidney stone disease. Advances in Applied Microbiology 72: 63–87. [Google Scholar]
Adams DH, Eksteen B. (2006). Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nature Reviews Immunology 6: 244–251. DOI 10.1038/nri1784. [Google Scholar] [CrossRef]
Ahn C, Negus D, Huang W. (2018). Pyoderma gangrenosum: A review of pathogenesis and treatment. Expert Review of Clinical Immunology 14: 225–233. DOI 10.1080/1744666X.2018.1438269. [Google Scholar] [CrossRef]
Ambruzs JM, Walker PD, Larsen CP. (2014). The histopathologic spectrum of kidney biopsies in patients with inflammatory bowel disease. Clinical Journal of the American Society of Nephrology 9: 265–270. DOI 10.2215/CJN.04660513. [Google Scholar] [CrossRef]
Anders HJ, Banas B, Schlöndorff D (2004a). Signaling danger: Toll-like receptors and their potential roles in kidney disease. Journal of the American Society of Nephrology 15: 854–867. DOI 10.1097/01.ASN.0000121781.89599.16. [Google Scholar] [CrossRef]
Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, Perez de Lema G, Schlöndorff D (2004b). Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB Journal 18: 534–536. DOI 10.1096/fj.03-0646fje. [Google Scholar] [CrossRef]
Aniwan S, Pardi DS, Tremaine WJ, Loftus EVJr. (2018). Increased risk of acute myocardial infarction and heart failure in patients with inflammatory bowel diseases. Clinical Gastroenterology and Hepatology 16: 1607–1615.e1. DOI 10.1016/j.cgh.2018.04.031. [Google Scholar] [CrossRef]
Annese V. (2019). A review of extraintestinal manifestations and complications of inflammatory bowel disease. Saudi Journal of Medicine and Medical Sciences 7: 66–73. DOI 10.4103/sjmms.sjmms_81_18. [Google Scholar] [CrossRef]
Arvans D, Jung YC, Antonopoulos D, Koval J, Granja I, Bashir M, Hassan H. (2017). Oxalobacter formigenes– derived bioactive factors stimulate oxalate transport by intestinal epithelial cells. Journal of the American Society of Nephrology 28: 876–887. DOI 10.1681/ASN.2016020132. [Google Scholar] [CrossRef]
Arvikar SL, Fisher MC. (2011). Inflammatory bowel disease associated arthropathy. Current Reviews in Musculoskeletal Medicine 4: 123–131. DOI 10.1007/s12178-011-9085-8. [Google Scholar] [CrossRef]
Asquith M, Elewaut D, Lin P, Rosenbaum JT. (2014). The role of the gut and microbes in the pathogenesis of spondyloarthritis. Best Practice & Research in Clinical Rheumatology 28: 687–702. DOI 10.1016/j.berh.2014.10.018. [Google Scholar] [CrossRef]
Bailey MT, Engler H, Sheridan JF. (2006). Stress induces the translocation of cutaneous and gastrointestinal microflora to secondary lymphoid organs of C57BL/6 mice. Journal of Neuroimmunology 171: 29–37. DOI 10.1016/j.jneuroim.2005.09.008. [Google Scholar] [CrossRef]
Bajer L, Kverka M, Kostovcik M, Macinga P, Dvorak J, Stehlikova Z, Drastich P. (2017). Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World Journal of Gastroenterology 23: 4548–4558. DOI 10.3748/wjg.v23.i25.4548. [Google Scholar] [CrossRef]
Banati M, Csecsei P, Koszegi E, Nielsen HH, Suto G, Bors L, Illes Z. (2013). Antibody response against gastrointestinal antigens in demyelinating diseases of the central nervous system. European Journal of Neurology 20: 1492–1495. [Google Scholar]
Beckman JS, Koppenol WH. (1996). Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. American Journal of Physiology-Cell Physiology 271: C1424–C1437. DOI 10.1152/ajpcell.1996.271.5.C1424. [Google Scholar] [CrossRef]
Bellanti F, Villani R, Facciorusso A, Vendemiale G, Serviddio G. (2017). Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radical Biology and Medicine 111: 173–185. DOI 10.1016/j.freeradbiomed.2017.01.023. [Google Scholar] [CrossRef]
Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Lusis AJ. (2013). Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metabolism 17: 49–60. DOI 10.1016/j.cmet.2012.12.011. [Google Scholar] [CrossRef]
Benstoem C, Goetzenich A, Kraemer S, Borosch S, Manzanares W, Hardy G, Stoppe C. (2015). Selenium and its supplementation in cardiovascular disease--what do we know? Nutrients 7: 3094–3118. DOI 10.3390/nu7053094. [Google Scholar] [CrossRef]
Bernstein CN. (2017). The brain-gut axis and stress in inflammatory bowel disease. Gastroenterology Clinics of North America 46: 839–846. DOI 10.1016/j.gtc.2017.08.006. [Google Scholar] [CrossRef]
Bianchi L, Gaiani F, Vincenzi F, Kayali S, Di Mario F, Leandro G, Ruberto C. (2018). Hemolytic uremic syndrome: Differential diagnosis with the onset of inflammatory bowel diseases. Acta Biomedica 89: 153–157. [Google Scholar]
Bigeh A, Sanchez A, Maestas C, Gulati M. (2019). Inflammatory bowel disease and the risk for cardiovascular disease: Does all inflammation lead to heart disease? Trends in Cardiovascular Medicine 30: 463–469. DOI 10.1016/j.tcm.2019.10.001. [Google Scholar] [CrossRef]
Binion DG, Fu S, Ramanujam KS, Chai YC, Dweik RA, Drazba JA, Wilson KT. (1998). iNOS expression in human intestinal microvascular endothelial cells inhibits leukocyte adhesion. American Journal of Physiology 275: G592–603. [Google Scholar]
Binion DG, Rafiee P, Ramanujam KS, Fu S, Fisher PJ, Rivera MT, Wilson KT. (2000). Deficient iNOS in inflammatory bowel disease intestinal microvascular endothelial cells results in increased leukocyte adhesion. Free Radical Biology and Medicine 29: 881–888. DOI 10.1016/S0891-5849(00)00391-9. [Google Scholar] [CrossRef]
Block ML, Zecca L, Hong JS. (2007). Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nature Reviews Neuroscience 8: 57–69. DOI 10.1038/nrn2038. [Google Scholar] [CrossRef]
Brakenhoff LK, van der Heijde DM, Hommes DW, Huizinga TW, Fidder HH. (2010). The joint–gut axis in inflammatory bowel diseases. Journal of Crohn’s and Colitis 4: 257–268. DOI 10.1016/j.crohns.2009.11.005. [Google Scholar] [CrossRef]
Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, Korecka A, Bakocevic N, Guan Ng L, Kundu G, Gulyás B, Halldin C, Hultenby K, Nilsson H, Hebert H, Volpe BT, Diamond B, Pettersson S. (2014). The gut microbiota influences blood-brain barrier permeability in mice. Science Translational Medicine 6: 263ra158. DOI 10.1126/scitranslmed.3009759. [Google Scholar] [CrossRef]
Campieri C, Campieri M, Bertuzzi V, Swennen E, Matteuzzi D, Stefoni S, Pirovano F, Centi C, Ulisse S, Famularo G, De Simone C. (2001). Reduction of oxaluria after an oral course of lactic acid bacteria at high concentration. Kidney International 60: 1097–1105. DOI 10.1046/j.1523-1755.2001.0600031097.x. [Google Scholar] [CrossRef]
Cao LC, Honeyman T, Jonassen J, Scheid C. (2000). Oxalate-induced ceramide accumulation in Madin-Darby canine kidney and LLC-PK1 cells. Kidney International 57: 2403–2411. DOI 10.1046/j.1523-1755.2000.00099.x. [Google Scholar] [CrossRef]
Cao LC, Honeyman TW, Cooney R, Kennington L, Scheid CR, Jonassen JA. (2004). Mitochondrial dysfunction is a primary event in renal cell oxalate toxicity. Kidney International 66: 1890–1900. DOI 10.1111/j.1523-1755.2004.00963.x. [Google Scholar] [CrossRef]
Capon F, Di Meglio P, Szaub J, Prescott NJ, Dunster C, Baumber L, Nestle FO. (2007). Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Human Genetics 122: 201–206. DOI 10.1007/s00439-007-0397-0. [Google Scholar] [CrossRef]
Casella G, Tontini GE, Bassotti G, Pastorelli L, Villanacci V, Spina L, Vecchi M. (2014). Neurological disorders and inflammatory bowel diseases. World Journal of Gastroenterology 20: 8764–8782. [Google Scholar]
Castro Aguilar-Tablada T, Navarro-Alarcon M, Quesada Granados J, Samaniego Sanchez C, Rufian-Henares JA, Nogueras-Lopez F. (2016). Ulcerative colitis and crohn’s disease are associated with decreased serum selenium concentrations and increased cardiovascular risk. Nutrients 8: 12. DOI 10.3390/nu8120780. [Google Scholar] [CrossRef]
Chakravarthy KS, Jayasudha R, Sai Prashanthi G, Ali MH, Sharma S, Tyagi M, Shivaji S. (2018). Dysbiosis in the gut bacterial microbiome of patients with uveitis, an inflammatory disease of the eye. Indian Journal of Microbiology 58: 457–469. DOI 10.1007/s12088-018-0746-9. [Google Scholar] [CrossRef]
Chao CY, Battat R, Al Khoury A, Restellini S, Sebastiani G, Bessissow T. (2016). Co-existence of non-alcoholic fatty liver disease and inflammatory bowel disease: A review article. World Journal of Gastroenterology 22: 7727–7734. DOI 10.3748/wjg.v22.i34.7727. [Google Scholar] [CrossRef]
Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B. (2010). Diagnosis and management of primary sclerosing cholangitis. American Association for the Study of Liver 51: 660–678. [Google Scholar]
Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. (2017). Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. Journal of the American Heart Association 6: 9. [Google Scholar]
Chen S, Li Y, Li S, Yu C. (2008). A Val227Ala substitution in the peroxisome proliferator activated receptor alpha (PPAR alpha) gene associated with non-alcoholic fatty liver disease and decreased waist circumference and waist-to-hip ratio. Journal of Gastroenterology and Hepatology 23: 1415–1418. DOI 10.1111/j.1440-1746.2008.05523.x. [Google Scholar] [CrossRef]
Chen X, Zhang Z, Li H, Zhao J, Wei X, Lin W, Zhao X, Jiang A, Yuan J. (2020). Endogenous ethanol produced by intestinal bacteria induces mitochondrial dysfunction in non-alcoholic fatty liver disease. Journal of Gastroenterology and Hepatology 35: 2009–2019. DOI 10.1111/jgh.15027. [Google Scholar] [CrossRef]
Cho JH. (2008). The genetics and immunopathogenesis of inflammatory bowel disease. Nature Reviews Immunology 8: 458–466. DOI 10.1038/nri2340. [Google Scholar] [CrossRef]
Ciccacci C, Biancone L, Di Fusco D, Ranieri M, Condino G, Giardina E, Onali S, Lepre T, Pallone F, Novelli G, Borgiani P. (2013). TRAF3IP2 gene is associated with cutaneous extraintestinal manifestations in inflammatory bowel disease. Journal of Crohn’s and Colitis 7: 44–52. DOI 10.1016/j.crohns.2012.02.020. [Google Scholar] [CrossRef]
Ciccia F, Ferrante A, Triolo G. (2016). Intestinal dysbiosis and innate immune responses in axial spondyloarthritis. Current Opinion in Rheumatology 28: 352–358. DOI 10.1097/BOR.0000000000000296. [Google Scholar] [CrossRef]
Cioffi M, Rosa AD, Serao R, Picone I, Vietri MT. (2015). Laboratory markers in ulcerative colitis: Current insights and future advances. World Journal of Gastrointestinal Pathophysiology 6: 13–22. DOI 10.4291/wjgp.v6.i1.13. [Google Scholar] [CrossRef]
Corica D, Romano C. (2016). Renal involvement in inflammatory bowel diseases. Journal of Crohn’s and Colitis 10: 226–235. DOI 10.1093/ecco-jcc/jjv138. [Google Scholar] [CrossRef]
Costello ME, Ciccia F, Willner D, Warrington N, Robinson PC, Gardiner B, Marshall M, Kenna TJ, Triolo G, Brown MA. (2015). Brief report: Intestinal dysbiosis in ankylosing spondylitis. Arthritis & Rheumatology 67: 686–691. DOI 10.1002/art.38967. [Google Scholar] [CrossRef]
Cottone M, Sapienza C, Macaluso FS, Cannizzaro M. (2019). Psoriasis and inflammatory bowel disease. Digestive Diseases 37: 451–457. DOI 10.1159/000500116. [Google Scholar] [CrossRef]
Danese S. (2007). Inflammation and the mucosal microcirculation in inflammatory bowel disease: The ebb and flow. Current Opinion in Gastroenterology 23: 384–389. DOI 10.1097/MOG.0b013e32810c8de3. [Google Scholar] [CrossRef]
Danese S, Papa A, Saibeni S, Repici A, Malesci A, Vecchi M. (2007). Inflammation and coagulation in inflammatory bowel disease: The clot thickens. American Journal of Gastroenterology 102: 174–186. DOI 10.1111/j.1572-0241.2006.00943.x. [Google Scholar] [CrossRef]
Davis C, Fischer J, Ley K, Sarembock IJ. (2003). The role of inflammation in vascular injury and repair. Journal of Thrombosis and Haemostasis 1: 1699–1709. DOI 10.1046/j.1538-7836.2003.00292.x. [Google Scholar] [CrossRef]
de La Serre CB, de Lartigue G, Raybould HE. (2015). Chronic exposure to low dose bacterial lipopolysaccharide inhibits leptin signaling in vagal afferent neurons. Physiology & Behavior 139: 188–194. DOI 10.1016/j.physbeh.2014.10.032. [Google Scholar] [CrossRef]
Del Rio D, Zimetti F, Caffarra P, Tassotti M, Bernini F, Brighenti F, Zini A, Zanotti I. (2017). The gut microbial metabolite trimethylamine-N-oxide is present in human cerebrospinal fluid. Nutrients 9: 10. DOI 10.3390/nu9101053. [Google Scholar] [CrossRef]
Demetter P, Baeten D, De Keyser F, De Vos M, Van Damme N, Verbruggen G, Vermeulen S, Mareel M, Elewaut G, Mielants H, Veys EM, Cuvelier CA. (2000). Subclinical gut inflammation in spondyloarthropathy patients is associated with upregulation of the E cadherin/catenin complex. Annals of the Rheumatic Diseases 59: 211–216. DOI 10.1136/ard.59.3.211. [Google Scholar] [CrossRef]
Demetter P, De Vos M, Van Huysse JA, Baeten D, Ferdinande L, Peeters H, Mielants H, Veys EM, De Keyser F, Cuvelier CA. (2005). Colon mucosa of patients both with spondyloarthritis and Crohn’s disease is enriched with macrophages expressing the scavenger receptor CD163. Annals of the Rheumatic Diseases 64: 321–324. DOI 10.1136/ard.2003.018382. [Google Scholar] [CrossRef]
Dijkstra G, Moshage H, van Dullemen HM, de Jager-Krikken A, Tiebosch AT, Kleibeuker GH, Jansen PL, van Goor H. (1998). Expression of nitric oxide synthases and formation of nitrotyrosine and reactive oxygen species in inflammatory bowel disease. Journal of Pathology 186: 416–421. [Google Scholar]
Drzewoski J, Gasiorowska A, Malecka-Panas E, Bald E, Czupryniak L. (2006). Plasma total homocysteine in the active stage of ulcerative colitis. Journal of Gastroenterology and Hepatology 21: 739–743. DOI 10.1111/j.1440-1746.2006.04255.x. [Google Scholar] [CrossRef]
Eaton JE, Talwalkar JA. (2013). Primary sclerosing cholangitis: Current and future management strategies. Current Hepatitis Reports 12: 28–36. DOI 10.1007/s11901-012-0155-1. [Google Scholar] [CrossRef]
Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hübscher SG, Briskin M, Salmon M, Adams DH. (2004). Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. Journal of Experimental Medicine 200: 1511–1517. DOI 10.1084/jem.20041035. [Google Scholar] [CrossRef]
El Kebir D, Jozsef L, Filep JG. (2008). Neutrophil recognition of bacterial DNA and Toll-like receptor 9-dependent and -independent regulation of neutrophil function. Archivum Immunologiae et Therapiae Experimentalis 56: 41–53. DOI 10.1007/s00005-008-0008-3. [Google Scholar] [CrossRef]
Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A, Debrus S, Raelson JV, Tejasvi T, Belouchi M, West SL, Barker JN, Kõks S, Kingo KC, Balschun T, Palmieri O, Annese V, Gieger C, Wichmann HE, Kabesch M, Trembath RC, Mathew CG, Abecasis GR, Weidinger S, Nikolaus S, Schreiber S, Elder JT, Weichenthal M, Nothnagel M, Franke A. (2012). Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. American Journal of Human Genetics 90: 636–647. DOI 10.1016/j.ajhg.2012.02.020. [Google Scholar] [CrossRef]
Elsehety A, Bertorini TE. (1997). Neurologic and neuropsychiatric complications of Crohn’s disease. Southern Medical Journal 90: 606–610. DOI 10.1097/00007611-199706000-00005. [Google Scholar] [CrossRef]
Eppinga H, Sperna Weiland CJ, Thio HB, van der Woude CJ, Nijsten TE, Peppelenbosch MP, Konstantinov SR. (2016). Similar depletion of protective Faecalibacterium prausnitzii in psoriasis and inflammatory bowel disease, but not in hidradenitis suppurativa. Journal of Crohn’s and Colitis 10: 1067–1075. DOI 10.1093/ecco-jcc/jjw070. [Google Scholar] [CrossRef]
Esmon CT. (2005). The interactions between inflammation and coagulation. British Journal of Haematology 131: 417–430. DOI 10.1111/j.1365-2141.2005.05753.x. [Google Scholar] [CrossRef]
Fantini MC, Pallone F, Monteleone G. (2009). Common immunologic mechanisms in inflammatory bowel disease and spondylarthropathies. World Journal of Gastroenterology 15: 2472–2478. DOI 10.3748/wjg.15.2472. [Google Scholar] [CrossRef]
Farhi D, Cosnes J, Zizi N, Chosidow O, Seksik P, Beaugerie L, Aractingi S, Khosrotehrani K. (2008). Significance of erythema nodosum and pyoderma gangrenosum in inflammatory bowel diseases: A cohort study of 2402 patients. Medicine 87: 281–293. DOI 10.1097/MD.0b013e318187cc9c. [Google Scholar] [CrossRef]
Feuerstein JD, Cheifetz AS. (2014). Ulcerative colitis: Epidemiology, diagnosis, and management. Mayo Clinic Proceedings 89: 1553–1563. DOI 10.1016/j.mayocp.2014.07.002. [Google Scholar] [CrossRef]
Floege J, Ketteler M. (2004). Vascular calcification in patients with end-stage renal disease. Nephrology Dialysis Transplantation 19: V59–66. DOI 10.1093/ndt/gfh1058. [Google Scholar] [CrossRef]
Forshaw MJ, Guirguis O, Hennigan TW. (2005). IgA nephropathy in association with Crohn’s disease. International Journal of Colorectal Disease 20: 463–465. DOI 10.1007/s00384-004-0696-z. [Google Scholar] [CrossRef]
Foster JA, McVey Neufeld KA. (2013). Gut–brain axis: How the microbiome influences anxiety and depression. Trends in Neuroscience 36: 305–312. DOI 10.1016/j.tins.2013.01.005. [Google Scholar] [CrossRef]
Fousekis FS, Theopistos VI, Katsanos KH, Tsianos EV, Christodoulou DK. (2018). Hepatobiliary manifestations and complications in inflammatory bowel disease: A review. Gastroenterology Research 11: 83–94. DOI 10.14740/gr990w. [Google Scholar] [CrossRef]
Fragoulis GE, Liava C, Daoussis D, Akriviadis E, Garyfallos A, Dimitroulas T. (2019). Inflammatory bowel diseases and spondyloarthropathies: From pathogenesis to treatment. World Journal of Gastroenterology 25: 2162–2176. DOI 10.3748/wjg.v25.i18.2162. [Google Scholar] [CrossRef]
Fraser JS, Muller AF, Smith DJ, Newman DJ, Lamb EJ. (2001). Renal tubular injury is present in acute inflammatory bowel disease prior to the introduction of drug therapy. Alimentary Pharmacology & Therapeutics 15: 1131–1137. DOI 10.1046/j.1365-2036.2001.01041.x. [Google Scholar] [CrossRef]
Fried L, Solomon C, Shlipak M, Seliger S, Stehman-Breen C, Bleyer AJ, Chaves P, Furberg C, Kuller L, Newman A. (2004). Inflammatory and prothrombotic markers and the progression of renal disease in elderly individuals. Journal of the American Society of Nephrology 15: 3184–3191. DOI 10.1097/01.ASN.0000146422.45434.35. [Google Scholar] [CrossRef]
Fu Y, Lee CH, Chi CC. (2018). Association of psoriasis with inflammatory bowel disease: A systematic review and meta-analysis. JAMA Dermatology 154: 1417–1423. DOI 10.1001/jamadermatol.2018.3631. [Google Scholar] [CrossRef]
Fujimoto T, Imaeda H, Takahashi K, Kasumi E, Bamba S, Fujiyama Y, Andoh A. (2013). Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. Journal of Gastroenterology and Hepatology 28: 613–619. DOI 10.1111/jgh.12073. [Google Scholar] [CrossRef]
Ganji-Arjenaki M, Nasri H, Rafieian-Kopaei M. (2017). Nephrolithiasis as a common urinary system manifestation of inflammatory bowel diseases; a clinical review and meta-analysis. Journal of Nephropathology 6: 264–269. DOI 10.15171/jnp.2017.42. [Google Scholar] [CrossRef]
Garber A, Regueiro M. (2019). Extraintestinal manifestations of inflammatory bowel disease: Epidemiology, etiopathogenesis, and management. Current Gastroenterology Reports 21: 31. DOI 10.1007/s11894-019-0698-1. [Google Scholar] [CrossRef]
Gilis E, Mortier C, Venken K, Debusschere K, Vereecke L, Elewaut D. (2018). The role of the microbiome in gut and joint inflammation in psoriatic arthritis and spondyloarthritis. Journal of Rheumatology Supplement 94: 36–39. [Google Scholar]
Gionchetti P, Calabrese C, Rizzello F. (2015). Inflammatory Bowel Diseases and Spondyloarthropathies. The Journal of Rheumatology Supplement 93: 21–23. DOI 10.3899/jrheum.150628. [Google Scholar] [CrossRef]
Gkentzis A, Kimuli M, Cartledge J, Traxer O, Biyani CS. (2016). Urolithiasis in inflammatory bowel disease and bariatric surgery. World Journal of Nephrology 5: 538–546. DOI 10.5527/wjn.v5.i6.538. [Google Scholar] [CrossRef]
Gondim FA, Brannagan TH, III, Sander HW, Chin RL, Latov N. (2005). Peripheral neuropathy in patients with inflammatory bowel disease. Brain 128: 867–879. DOI 10.1093/brain/awh429. [Google Scholar] [CrossRef]
Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH. (2002). Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet 359: 150–157. DOI 10.1016/S0140-6736(02)07374-9. [Google Scholar] [CrossRef]
Gray MT, Woulfe JM. (2015). Striatal blood–brain barrier permeability in Parkinson’s disease. Journal of Cerebral Blood Flow & Metabolism 35: 747–750. DOI 10.1038/jcbfm.2015.32. [Google Scholar] [CrossRef]
Green S, Wahli W. (1994). Peroxisome proliferator-activated receptors: Finding the orphan a home. Molecular and Cellular Endocrinology 100: 149–153. DOI 10.1016/0303-7207(94)90294-1. [Google Scholar] [CrossRef]
Greuter T, Vavricka SR. (2019). Extraintestinal manifestations in inflammatory bowel disease–epidemiology, genetics, and pathogenesis. Expert Review of Gastroenterology & Hepatology 13: 307–317. DOI 10.1080/17474124.2019.1574569. [Google Scholar] [CrossRef]
Gupta G, Gelfand JM, Lewis JD. (2005). Increased risk for demyelinating diseases in patients with inflammatory bowel disease. Gastroenterology 129: 819–826. DOI 10.1053/j.gastro.2005.06.022. [Google Scholar] [CrossRef]
Hansen TH, Gobel RJ, Hansen T, Pedersen O. (2015). The gut microbiome in cardio-metabolic health. Genome Medicine 7: 33. DOI 10.1186/s13073-015-0157-z. [Google Scholar] [CrossRef]
Hansson GK. (2005). Inflammation, atherosclerosis, and coronary artery disease. New England Journal of Medicine 353: 429–430, author reply 429–430. [Google Scholar]
Harbord M, Annese V, Vavricka SR, Allez M, Barreiro-de Acosta M, Boberg KM, Colitis O. (2016). The first European evidence-based consensus on extra-intestinal manifestations in inflammatory bowel disease. Journal of Crohn’s and Colitis 10: 239–254. DOI 10.1093/ecco-jcc/jjv213. [Google Scholar] [CrossRef]
Hart A L, Al-Hassi HO, Rigby RJ, Bell SJ, Emmanuel AV, Knight SC, Kamm MA, Stagg AJ. (2005). Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology 129: 50–65. DOI 10.1053/j.gastro.2005.05.013. [Google Scholar] [CrossRef]
Hatoum OA, Binion DG, Otterson MF, Gutterman DD. (2003). Acquired microvascular dysfunction in inflammatory bowel disease: Loss of nitric oxide-mediated vasodilation. Gastroenterology 125: 58–69. DOI 10.1016/S0016-5085(03)00699-1. [Google Scholar] [CrossRef]
Hermann E, Yu DT, Meyer zum Buschenfelde KH, Fleischer B. (1993). HLA-B27-restricted CD8 T cells derived from synovial fluids of patients with reactive arthritis and ankylosing spondylitis. Lancet 342: 646–650. DOI 10.1016/0140-6736(93)91760-J. [Google Scholar] [CrossRef]
Higa A, Chevet E. (2012). Redox signaling loops in the unfolded protein response. Cellular Signalling 24: 1548–1555. DOI 10.1016/j.cellsig.2012.03.011. [Google Scholar] [CrossRef]
Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. (2013). Primary sclerosing cholangitis. Lancet 382: 1587–1599. DOI 10.1016/S0140-6736(13)60096-3. [Google Scholar] [CrossRef]
Hoban DB, Connaughton E, Connaughton C, Hogan G, Thornton C, Mulcahy P, Moloney T C, Dowd E. (2013). Further characterisation of the LPS model of Parkinson’s disease: A comparison of intra-nigral and intra-striatal lipopolysaccharide administration on motor function, microgliosis and nigrostriatal neurodegeneration in the rat. Brain, Behavior, and Immunity 27: 91–100. DOI 10.1016/j.bbi.2012.10.001. [Google Scholar] [CrossRef]
Horai R, Caspi RR. (2019). Microbiome and autoimmune uveitis. Frontiers in Immunology 10: 232. DOI 10.3389/fimmu.2019.00232. [Google Scholar] [CrossRef]
Horai R, Zarate-Blades CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, Jittayasothorn Y, Chan CC, Yamane H, Honda K, Caspi RR. (2015). Microbiota-dependent activation of an autoreactive T cell receptor provokes autoimmunity in an immunologically privileged site. Immunity 43: 343–353. DOI 10.1016/j.immuni.2015.07.014. [Google Scholar] [CrossRef]
Horowitz S, Binion DG, Nelson VM, Kanaa Y, Javadi P, Lazarova Z, Andrekopoulos C, Kalyanaraman B, Otterson MF, Rafiee P. (2007). Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease. American Journal of Physiology-Gastrointestinal and Liver Physiology 292: G1323–G1336. DOI 10.1152/ajpgi.00499.2006. [Google Scholar] [CrossRef]
Howell KW, Meng X, Fullerton DA, Jin C, Reece TB, Cleveland JCJr. (2011). Toll-like receptor 4 mediates oxidized LDL-induced macrophage differentiation to foam cells. Journal of Surgical Research 171: e27–31. DOI 10.1016/j.jss.2011.06.033. [Google Scholar] [CrossRef]
Huang X, Ye Z, Cao Q, Su G, Wang Q, Deng J, Zhou C, Kijlstra A, Yang P. (2018). Gut microbiota composition and fecal metabolic phenotype in patients with acute anterior uveitis. Investigative Opthalmology & Visual Science 59: 1523–1531. DOI 10.1167/iovs.17-22677. [Google Scholar] [CrossRef]
Hudson M, Hutton RA, Wakefield AJ, Sawyerr AM, Pounder RE. (1992). Evidence for activation of coagulation in Crohn’s disease. Blood Coagulation & Fibrinolysis 3: 773–778. DOI 10.1097/00001721-199212000-00011. [Google Scholar] [CrossRef]
Hueppelshaeuser R, von Unruh GE, Habbig S, Beck BB, Buderus S, Hesse A, Hoppe B. (2012). Enteric hyperoxaluria, recurrent urolithiasis, and systemic oxalosis in patients with Crohn’s disease. Pediatric Nephrology 27: 1103–1109. DOI 10.1007/s00467-012-2126-8. [Google Scholar] [CrossRef]
Hussein IAH, Tohme R, Barada K, Mostafa MH, Freund JN, Jurjus RA, Karam W, Jurjus A. (2008). Inflammatory bowel disease in rats: Bacterial and chemical interaction. World Journal of Gastroenterology 14: 4028–4039. DOI 10.3748/wjg.14.4028. [Google Scholar] [CrossRef]
Jacques P, Elewaut D. (2008). Joint expedition: Linking gut inflammation to arthritis. Mucosal Immunology 1: 364–371. DOI 10.1038/mi.2008.24. [Google Scholar] [CrossRef]
Janowitz C, Nakamura YK, Metea C, Gligor A, Yu W, Karstens L, Rosenbaum JT, Asquith M, Lin P. (2019). Disruption of intestinal homeostasis and intestinal microbiota during experimental autoimmune uveitis. Investigative Opthalmology & Visual Science 60: 420–429. DOI 10.1167/iovs.18-24813. [Google Scholar] [CrossRef]
Janse M, Lamberts LE, Franke L, Raychaudhuri S, Ellinghaus E, Muri Boberg K, Melum E, Folseraas T, Schrumpf E, Bergquist A, Björnsson E, Fu J, Jan Westra H, Groen HJM, Fehrmann RSN, Smolonska J, van den Berg LH, Ophoff RA, Porte RJ, Weismüller TJ, Wedemeyer J, Schramm C, Sterneck M, Günther R, Braun F, Vermeire S, Henckaerts L, Wijmenga C, Ponsioen CY, Schreiber S, Karlsen TH, Franke A, Weersma RK. (2011). Three ulcerative colitis susceptibility loci are associated with primary sclerosing cholangitis and indicate a role for IL2, REL, and CARD9. Hepatology 53: 1977–1985. DOI 10.1002/hep.24307. [Google Scholar] [CrossRef]
Jonassen JA, Kohjimoto Y, Scheid CR, Schmidt M. (2005). Oxalate toxicity in renal cells. Urological Research 33: 329–339. DOI 10.1007/s00240-005-0485-3. [Google Scholar] [CrossRef]
Jorens PG, Hermans CR, Haber I, Kockx MM, Vermylen J, Parizel GA. (1990). Acquired protein C and S deficiency, inflammatory bowel disease and cerebral arterial thrombosis. Blut 61: 307–310. DOI 10.1007/BF01732883. [Google Scholar] [CrossRef]
Joseph L, Fink LM, Hauer-Jensen M. (2002). Cytokines in coagulation and thrombosis: A preclinical and clinical review. Blood Coagulation & Fibrinolysis 13: 105–116. DOI 10.1097/00001721-200203000-00005. [Google Scholar] [CrossRef]
Kamada N, Núñez G. (2013). Role of the gut microbiota in the development and function of lymphoid cells. Journal of Immunology 190: 1389–1395. DOI 10.4049/jimmunol.1203100. [Google Scholar] [CrossRef]
Kane S. (2006). Urogenital complications of Crohn’s disease. American Journal of Gastroenterology 101: S640–643. DOI 10.1111/j.1572-0241.2006.00662.x. [Google Scholar] [CrossRef]
Karlsen TH, Franke A, Melum E, Kaser A, Hov JR, Balschun T, Lie BA, Bergquist A, Schramm C, Weismüller TJ, Gotthardt D, Rust C, Philipp EER, Fritz T, Henckaerts L, Weersma RK, Stokkers P, Ponsioen CY, Wijmenga C, Sterneck M, Nothnagel M, Hampe J, Teufel A, Runz H, Rosenstiel P, Stiehl A, Vermeire S, Beuers U, Manns MP, Schrumpf E, Boberg KM, Schreiber S. (2010). Genome-wide association analysis in primary sclerosing cholangitis. Gastroenterology 138: 1102–1111. DOI 10.1053/j.gastro.2009.11.046. [Google Scholar] [CrossRef]
Karreman MC, Luime JJ, Hazes JMW, Weel A. (2017). The prevalence and incidence of axial and peripheral spondyloarthritis in inflammatory bowel disease: A systematic review and meta-analysis. Journal of Crohn’s and Colitis 11: 631–642. [Google Scholar]
Kaur CP, Vadivelu J, Chandramathi S. (2018). Impact of Klebsiella pneumoniae in lower gastrointestinal tract diseases. Journal of Digestive Diseases 19: 262–271. DOI 10.1111/1751-2980.12595. [Google Scholar] [CrossRef]
Knutson CG, Mangerich A, Zeng Y, Raczynski AR, Liberman RG, Kang P, Ye W, Prestwich EG, Lu K, Wishnok JS, Korzenik JR, Wogan GN, Fox JG, Dedon PC, Tannenbaum SR. (2013). Chemical and cytokine features of innate immunity characterize serum and tissue profiles in inflammatory bowel disease. Proceedings of the National Academy of Sciences of the United States of America 10: 2332–2341. DOI 10.1073/pnas.1222669110. [Google Scholar] [CrossRef]
Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WHW, Bushman FD, Lusis AJ, Hazen SL. (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature Medicine 19: 576–585. DOI 10.1038/nm.3145. [Google Scholar] [CrossRef]
Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Takayanagi R, Nakamuta M. (2007). Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. International Journal of Molecular Medicine 20: 351–358. [Google Scholar]
Kolios G, Valatas V, Ward SG. (2004). Nitric oxide in inflammatory bowel disease: A universal messenger in an unsolved puzzle. Immunology 113: 427–437. DOI 10.1111/j.1365-2567.2004.01984.x. [Google Scholar] [CrossRef]
Kumar R, Ghoshal UC, Singh G, Mittal RD. (2004). Infrequency of colonization with Oxalobacter formigenes in inflammatory bowel disease: Possible role in renal stone formation. Journal of Gastroenterology and Hepatology 19: 1403–1409. DOI 10.1111/j.1440-1746.2004.03510.x. [Google Scholar] [CrossRef]
Kurilshikov A, van den Munckhof ICL, Chen L, Bonder MJ, Schraa K, Rutten JHW, Riksen NP, de Graaf J, Oosting M, Sanna S, Joosten LAB, van der Graaf M, Brand T, Koonen DPY, van Faassen M, Xavier RJ, Kuipers F, Hofker MH, Wijmenga C, Netea MG, Zhernakova A, Fu J. (2019). Gut microbial associations to plasma metabolites linked to cardiovascular phenotypes and risk. Circulation Research 124: 1808–1820. DOI 10.1161/CIRCRESAHA.118.314642. [Google Scholar] [CrossRef]
Larsen S, Bendtzen K, Nielsen OH. (2010). Extraintestinal manifestations of inflammatory bowel disease: Epidemiology, diagnosis, and management. Annals of Medicine 42: 97–114. DOI 10.3109/07853890903559724. [Google Scholar] [CrossRef]
Lee IA, Kim DH. (2011). Klebsiella pneumoniae increases the risk of inflammation and colitis in a murine model of intestinal bowel disease. Scandinavian Journal of Gastroenterology 46: 684–693. DOI 10.3109/00365521.2011.560678. [Google Scholar] [CrossRef]
Leung TM, Nieto N. (2013). CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of Hepatology 58: 395–398. DOI 10.1016/j.jhep.2012.08.018. [Google Scholar] [CrossRef]
Levi M, van der Poll T, Schultz M. (2012). New insights into pathways that determine the link between infection and thrombosis. Netherlands Journal of Medicine 70: 114–120. [Google Scholar]
Liao L, Schneider KM, Galvez EJC, Frissen M, Marschall HU, Su H, Hatting M, Wahlström A, Haybaeck J, Puchas P, Mohs A, Peng J, Bergheim I, Nier A, Hennings J, Reißing J, Zimmermann HW, Longerich T, Strowig T, Liedtke C, Cubero FJ, Trautwein C. (2019). Intestinal dysbiosis augments liver disease progression via NLRP3 in a murine model of primary sclerosing cholangitis. Gut 68: 1477–1492. DOI 10.1136/gutjnl-2018-316670. [Google Scholar] [CrossRef]
Lin CH, Kadakia S, Frieri M (2014a). New insights into an autoimmune mechanism, pharmacological treatment and relationship between multiple sclerosis and inflammatory bowel disease. Autoimmunity Reviews 13: 114–116. DOI 10.1016/j.autrev.2013.09.011. [Google Scholar] [CrossRef]
Lin P, Bach M, Asquith M, Lee AY, Akileswaran L, Stauffer P, Davin S, Pan Y, Cambronne ED, Dorris M, Debelius JW, Lauber CL, Ackermann G, Baeza YV, Gill T, Knight R, Colbert RA, Taurog JD, Van Gelder RN, Rosenbaum JT, Bereswill S (2014b). HLA-B27 and human β2-microglobulin affect the gut microbiota of transgenic rats. PLoS One 9: e105684. DOI 10.1371/journal.pone.0105684. [Google Scholar] [CrossRef]
Liu HH, Shih TS, Huang HR, Huang SC, Lee LH, Huang YC. (2013). Plasma homocysteine is associated with increased oxidative stress and antioxidant enzyme activity in welders. Scientific World Journal 2013: 370487. DOI 10.1155/2013/370487. [Google Scholar] [CrossRef]
Liu M, Koh H, Kurtz ZD, Battaglia T, PeBenito A, Li H, Nazzal L, Blaser MJ. (2017). Oxalobacter formigenes-associated host features and microbial community structures examined using the American Gut Project. Microbiome 5: 108. DOI 10.1186/s40168-017-0316-0. [Google Scholar] [CrossRef]
Lossos A, River Y, Eliakim A, Steiner I. (1995). Neurologic aspects of inflammatory bowel disease. Neurology 45: 416–421. DOI 10.1212/WNL.45.3.416. [Google Scholar] [CrossRef]
Lyte M, Vulchanova L, Brown DR. (2011). Stress at the intestinal surface: Catecholamines and mucosa–bacteria interactions. Cell and Tissue Research 343: 23–32. DOI 10.1007/s00441-010-1050-0. [Google Scholar] [CrossRef]
Maes M, Berk M, Goehler L, Song C, Anderson G, Galecki P, Leonard B. (2012). Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Medicine 10: 66. DOI 10.1186/1741-7015-10-66. [Google Scholar] [CrossRef]
Maggs JR, Chapman RW. (2008). An update on primary sclerosing cholangitis. Current Opinion in Gastroenterology 24: 377–383. DOI 10.1097/MOG.0b013e3282f9e239. [Google Scholar] [CrossRef]
Magri S, Paduano D, Chicco F, Cingolani A, Farris C, Delogu G, Tumbarello F, Lai M, Melis A, Casula L, Fantini MC, Usai P. (2019). Nonalcoholic fatty liver disease in patients with inflammatory bowel disease: Beyond the natural history. World Journal of Gastroenterology 25: 5676–5686. DOI 10.3748/wjg.v25.i37.5676. [Google Scholar] [CrossRef]
Mahmud N, O’Toole D, O’Hare N, Freyne PJ, Weir DG, Kelleher D. (2002). Evaluation of renal function following treatment with 5-aminosalicylic acid derivatives in patients with ulcerative colitis. Alimentary Pharmacology & Therapeutics 16: 207–215. DOI 10.1046/j.1365-2036.2002.01155.x. [Google Scholar] [CrossRef]
Makrides SC. (1998). Therapeutic inhibition of the complement system. Pharmacological Reviews 50: 59–87. [Google Scholar]
Manichanh C, Borruel N, Casellas F, Guarner F. (2012). The gut microbiota in IBD. Nature Reviews Gastroenterology & Hepatology 9: 599–608. DOI 10.1038/nrgastro.2012.152. [Google Scholar] [CrossRef]
Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, García-Ruiz C. (2006). Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metabolism 4: 185–198. DOI 10.1016/j.cmet.2006.07.006. [Google Scholar] [CrossRef]
Maziere C, Conte MA, Dantin F, Maziere JC. (1999). Lipopolysaccharide enhances oxidative modification of low density lipoprotein by copper ions, endothelial and smooth muscle cells. Atherosclerosis 143: 75–80. DOI 10.1016/S0021-9150(98)00277-9. [Google Scholar] [CrossRef]
McAleer IM, Kaplan GW, Bradley JS, Carroll SF, Griffith DP. (2003). Endotoxin content in renal calculi. Journal of Urology 169: 1813–1814. DOI 10.1097/01.ju.0000061965.51478.79. [Google Scholar] [CrossRef]
McConnell N, Campbell S, Gillanders I, Rolton H, Danesh B. (2002). Risk factors for developing renal stones in inflammatory bowel disease. BJU International 89: 835–841. DOI 10.1046/j.1464-410X.2002.02739.x. [Google Scholar] [CrossRef]
Mehta M, Goldfarb DS, Nazzal L. (2016). The role of the microbiome in kidney stone formation. International Journal of Surgery 36: 607–612. DOI 10.1016/j.ijsu.2016.11.024. [Google Scholar] [CrossRef]
Mendes FD, Levy C, Enders FB, Loftus EVJr, Angulo P, Lindor KD. (2007). Abnormal hepatic biochemistries in patients with inflammatory bowel disease. American Journal of Gastroenterology 102: 344–350. DOI 10.1111/j.1572-0241.2006.00947.x. [Google Scholar] [CrossRef]
Mikocka-Walus A, Knowles SR, Keefer L, Graff L. (2016). Controversies revisited: A systematic review of the comorbidity of depression and anxiety with inflammatory bowel diseases. Inflammatory Bowel Disease 22: 752–762. DOI 10.1097/MIB.0000000000000620. [Google Scholar] [CrossRef]
Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. (2010). Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1β in mice. Gastroenterology 139: 323–334.e7. DOI 10.1053/j.gastro.2010.03.052. [Google Scholar] [CrossRef]
Moreira Mde L, Tsuji M, Corbett AJ, Araújo MSS, Teixeira-Carvalho A, Martins-Filho OA, Peruhype-Magalhães V, Coelho-dos-Reis JG. (2017). MAIT-cells: A tailor-made mate in the ancient battle against infectious diseases? Immunology Letters 187: 53–60. DOI 10.1016/j.imlet.2017.05.007. [Google Scholar] [CrossRef]
Moris G. (2014). Inflammatory bowel disease: An increased risk factor for neurologic complications. World Journal of Gastroenterology 20: 1228–1237. DOI 10.3748/wjg.v20.i5.1228. [Google Scholar] [CrossRef]
Moura FA, De Andrade KQ, Santos JCF, Araújo ORP, Goulart MOF. (2015). Antioxidant therapy for treatment of inflammatory bowel disease: Does it work? Redox Biology 6: 617–639. DOI 10.1016/j.redox.2015.10.006. [Google Scholar] [CrossRef]
Moura FA, de Andrade KD, Araújo ORP, Nunes-Souza V, Santos JCF, Rabelo LA, Goulart MD. (2016). Colonic and hepatic modulation by lipoic acid and/or N-acetylcysteine supplementation in mild ulcerative colitis induced by dextran sodium sulfate in rats. Oxidative Medicine and Cellular Longevity 2016: 4047362. DOI 10.1155/2016/4047362. [Google Scholar] [CrossRef]
Muriel P. (2009). Role of free radicals in liver diseases. Hepatology International 3: 526–536. DOI 10.1007/s12072-009-9158-6. [Google Scholar] [CrossRef]
Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. (2008). The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452: 103–107. DOI 10.1038/nature06664. [Google Scholar] [CrossRef]
Nakamoto N, Sasaki N, Aoki R, Miyamoto K, Suda W, Teratani T, Suzuki T, Koda Y, Chu PS, Taniki N, Yamaguchi A, Kanamori M, Kamada N, Hattori M, Ashida H, Sakamoto M, Atarashi K, Narushima S, Yoshimura A, Honda K, Sato T, Kanai T. (2019). Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nature Microbiology 4: 492–503. DOI 10.1038/s41564-018-0333-1. [Google Scholar] [CrossRef]
Nakamura YK, Metea C, Karstens L, Asquith M, Gruner H, Moscibrocki C, Lee I, Brislawn CJ, Jansson JK, Rosenbaum JT, Lin P. (2016). Gut microbial alterations associated with protection from autoimmune uveitis. Investigative Opthalmology & Visual Science 57: 3747–3758. DOI 10.1167/iovs.16-19733. [Google Scholar] [CrossRef]
Navaneethan U. (2014). Hepatobiliary manifestations of ulcerative colitis: An example of gut-liver crosstalk. Gastroenterology Reports 2: 193–200. DOI 10.1093/gastro/gou036. [Google Scholar] [CrossRef]
Nemati R, Mehdizadeh S, Salimipour H, Yaghoubi E, Alipour Z, Tabib SM, Assadi M. (2019). Neurological manifestations related to Crohn’s disease: A boon for the workforce. Gastroenterology Reports 7: 291–297. DOI 10.1093/gastro/gox034. [Google Scholar] [CrossRef]
Nguyen GC, Bernstein CN, Bitton A, Chan AK, Griffiths AM, Leontiadis GI, Geerts W, Bressler B, Butzner JD, Carrier M, Chande N, Marshall JK, Williams C, Kearon C. (2014). Consensus statements on the risk, prevention, and treatment of venous thromboembolism in inflammatory bowel disease: Canadian Association of Gastroenterology. Gastroenterology 146: 835–848.e6. DOI 10.1053/j.gastro.2014.01.042. [Google Scholar] [CrossRef]
Nielsen NM, Frisch M, Rostgaard K, Wohlfahrt J, Hjalgrim H, Koch-Henriksen N, Melbye M, Westergaard T. (2008). Autoimmune diseases in patients with multiple sclerosis and their first-degree relatives: A nationwide cohort study in Denmark. Multiple Sclerosis Journal 14: 823–829. DOI 10.1177/1352458508088936. [Google Scholar] [CrossRef]
Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. (2018). Gut microbiota in the pathogenesis of inflammatory bowel disease. Clinical Journal of Gastroenterology 11: 1–10. DOI 10.1007/s12328-017-0813-5. [Google Scholar] [CrossRef]
Nishino K, Nishida A, Inoue R, Kawada Y, Ohno M, Sakai S, Inatomi O, Bamba S, Sugimoto M, Kawahara M, Naito Y, Andoh A. (2018). Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. Journal of Gastroenterology 53: 95–106. DOI 10.1007/s00535-017-1384-4. [Google Scholar] [CrossRef]
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. (2000). Multiple sclerosis. New England Journal of Medicine 343: 938–952. DOI 10.1056/NEJM200009283431307. [Google Scholar] [CrossRef]
O’Mahony S, Anderson N, Nuki G, Ferguson A. (1992). Systemic and mucosal antibodies to Klebsiella in patients with ankylosing spondylitis and Crohn’s disease. Annals of the Rheumatic Diseases 51: 1296–1300. DOI 10.1136/ard.51.12.1296. [Google Scholar] [CrossRef]
Oikonomou K, Kapsoritakis A, Eleftheriadis T, Stefanidis I, Potamianos S. (2011). Renal manifestations and complications of inflammatory bowel disease. Inflammatory Bowel Diseases 17: 1034–1045. DOI 10.1002/ibd.21468. [Google Scholar] [CrossRef]
Olpin JD, Sjoberg BP, Stilwill SE, Jensen LE, Rezvani M, Shaaban AM. (2017). Beyond the bowel: Extraintestinal manifestations of inflammatory bowel disease. Radiographics 37: 1135–1160. DOI 10.1148/rg.2017160121. [Google Scholar] [CrossRef]
Orchard TR, Chua CN, Ahmad T, Cheng H, Welsh KI, Jewell DP. (2002). Uveitis and erythema nodosum in inflammatory bowel disease: Clinical features and the role of HLA genes. Gastroenterology 123: 714–718. DOI 10.1053/gast.2002.35396. [Google Scholar] [CrossRef]
Osna NA, Carter WG, Ganesan M, Kirpich IA, McClain CJ, Petersen DR, Shearn CT, Tomasi ML, Kharbanda KK. (2016). Aberrant post-translational protein modifications in the pathogenesis of alcohol-induced liver injury. World Journal of Gastroenterology 22: 6192–6200. DOI 10.3748/wjg.v22.i27.6192. [Google Scholar] [CrossRef]
Oussalah A, Gueant JL, Peyrin-Biroulet L. (2011). Meta-analysis: Hyperhomocysteinaemia in inflammatory bowel diseases. Alimentary Pharmacology & Therapeutics 34: 1173–1184. DOI 10.1111/j.1365-2036.2011.04864.x. [Google Scholar] [CrossRef]
Pagliassotti MJ. (2012). Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annual Review of Nutrition 32: 17–33. DOI 10.1146/annurev-nutr-071811-150644. [Google Scholar] [CrossRef]
Palumbo C, Restellini S, Chao CY, Aruljothy A, Lemieux C, Wild G, Afif W, Lakatos PL, Bitton A, Cocciolillo S, Ghali P, Bessissow T, Sebastiani G. (2019). Screening for nonalcoholic fatty liver disease in inflammatory bowel diseases: A cohort study using transient elastography. Inflammatory Bowel Diseases 25: 124–133. DOI 10.1093/ibd/izy200. [Google Scholar] [CrossRef]
Panes J, Granger DN. (1998). Leukocyte-endothelial cell interactions: Molecular mechanisms and implications in gastrointestinal disease. Gastroenterology 114: 1066–1090. DOI 10.1016/S0016-5085(98)70328-2. [Google Scholar] [CrossRef]
Papa A, Scaldaferri F, Danese S, Guglielmo S, Roberto I, Bonizzi M, Mocci G, Felice C, Ricci C, Andrisani G, Fedeli G, Gasbarrini G, Gasbarrini A. (2008). Vascular involvement in inflammatory bowel disease: Pathogenesis and clinical aspects. Digestive Diseases 26: 149–155. DOI 10.1159/000116773. [Google Scholar] [CrossRef]
Park S, Chun J, Han KD, Soh H, Choi K, Kim JH, Lee J, Lee C, Im JP, Kim JS. (2018). Increased end-stage renal disease risk in patients with inflammatory bowel disease: A nationwide population-based study. World Journal of Gastroenterology 24: 4798–4808. DOI 10.3748/wjg.v24.i42.4798. [Google Scholar] [CrossRef]
Parks JH, Worcester EM, O’Connor RC, Coe FL. (2003). Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney International 63: 255–265. DOI 10.1046/j.1523-1755.2003.00725.x. [Google Scholar] [CrossRef]
Pimentel-Santos FM, Matos M, Ligeiro D, Mourão AF, Ribeiro C, Costa J, Santos H, Barcelos A, Pinto P, Cruz M, Sousa E, Santos RA, Fonseca JE, Trindade H, Guedes-Pinto H, Branco JC. (2013). HLA alleles and HLA-B27 haplotypes associated with susceptibility and severity of ankylosing spondylitis in a Portuguese population. Tissue Antigens 82: 374–379. DOI 10.1111/tan.12238. [Google Scholar] [CrossRef]
Poulou AC, Goumas KE, Dandakis DC, Tyrmpas I, Panagiotaki M, Georgouli A, Soutos DC, Archimandritis A. (2006). Microproteinuria in patients with inflammatory bowel disease: Is it associated with the disease activity or the treatment with 5-aminosalicylic acid? World Journal of Gastroenterology 12: 739–746. DOI 10.3748/wjg.v12.i5.739. [Google Scholar] [CrossRef]
Primas C, Novacek G, Schweiger K, Mayer A, Eser A, Papay P, Gratzer C, Angelberger S, Dejaco C, Reinisch W, Vogelsang H. (2013). Renal insufficiency in IBD—prevalence and possible pathogenetic aspects. Journal of Crohn’s and Colitis 7: e630–e634. DOI 10.1016/j.crohns.2013.05.001. [Google Scholar] [CrossRef]
Purchiaroni F, Tortora A, Gabrielli M, Bertucci F, Gigante G, Ianiro G, Ojetti V, Scarpellini E, Gasbarrini A. (2013). The role of intestinal microbiota and the immune system. European Review for Medical and Pharmacological Sciences 17: 323–333. [Google Scholar]
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich S D, Wang J. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65. DOI 10.1038/nature08821. [Google Scholar] [CrossRef]
Qin L, Liu Y, Hong JS, Crews FT. (2013). NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia 61: 855–868. DOI 10.1002/glia.22479. [Google Scholar] [CrossRef]
Quevrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, Miquel S, Carlier L, Bermúdez-Humarán LG, Pigneur B, Lequin O, Kharrat P, Thomas G, Rainteau D, Aubry C, Breyner N, Afonso C, Lavielle S, Grill JP, Chassaing G, Chatel JM, Trugnan G, Xavier R, Langella P, Sokol H, Seksik P. (2016). Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut 65: 415–425. DOI 10.1136/gutjnl-2014-307649. [Google Scholar] [CrossRef]
Rahman I, Biswas SK. (2004). Non-invasive biomarkers of oxidative stress: Reproducibility and methodological issues. Redox Report 9: 125–143. DOI 10.1179/135100004225005219. [Google Scholar] [CrossRef]
Ratajczak AE, Szymczak-Tomczak A, Skrzypczak-Zielińska M, Rychter AM, Zawada A, Dobrowolska A, Krela-Kaźmierczak I. (2020). Vitamin C deficiency and the risk of osteoporosis in patients with an inflammatory bowel disease. Nutrients 12: 2263. DOI 10.3390/nu12082263. [Google Scholar] [CrossRef]
Restellini S, Chazouilleres O, Frossard JL. (2017). Hepatic manifestations of inflammatory bowel diseases. Liver International 37: 475–489. DOI 10.1111/liv.13265. [Google Scholar] [CrossRef]
Reyes-Gordillo K, Shah R, Muriel P. (2017). Oxidative stress and inflammation in hepatic diseases: Current and future therapy. Oxidative Medicine and Cellular Longevity 2017: 3140673. DOI 10.1155/2017/3140673. [Google Scholar] [CrossRef]
Robertson G, Leclercq I, Farrell GC. (2001). Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. American Journal of Physiology–Gastrointestinal and Liver Physiology 281: G1135–1139. DOI 10.1152/ajpgi.2001.281.5.G1135. [Google Scholar] [CrossRef]
Roifman I, Sun YC, Fedwick JP, Panaccione R, Buret AG, Liu H, Rostom A, Anderson TJ, Beck PL. (2009). Evidence of endothelial dysfunction in patients with inflammatory bowel disease. Clinical Gastroenterology and Hepatology 7: 175–182. DOI 10.1016/j.cgh.2008.10.021. [Google Scholar] [CrossRef]
Sabino J, Vieira-Silva S, Machiels K, Joossens M, Falony G, Ballet V, Ferrante M, Van Assche G, Van der Merwe S, Vermeire S, Raes J. (2016). Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 65: 1681–1689. DOI 10.1136/gutjnl-2015-311004. [Google Scholar] [CrossRef]
Sadaf H, Raza SI, Hassan SW. (2017). Role of gut microbiota against calcium oxalate. Microbial Pathogenesis 109: 287–291. DOI 10.1016/j.micpath.2017.06.009. [Google Scholar] [CrossRef]
Salmi M, Jalkanen S. (2001). Human leukocyte subpopulations from inflamed gut bind to joint vasculature using distinct sets of adhesion molecules. Journal of Immunology 166: 4650–4657. DOI 10.4049/jimmunol.166.7.4650. [Google Scholar] [CrossRef]
Santos J, Saunders PR, Hanssen NP, Yang PC, Yates D, Groot JA, Perdue MH. (1999). Corticotropin-releasing hormone mimics stress-induced colonic epithelial pathophysiology in the rat. American Journal of Physiology 277: G391–399. [Google Scholar]
Scaldaferri F, Lancellotti S, Pizzoferrato M, De Cristofaro R. (2011). Haemostatic system in inflammatory bowel diseases: New players in gut inflammation. World Journal of Gastroenterology 17: 594–608. DOI 10.3748/wjg.v17.i5.594. [Google Scholar] [CrossRef]
Scaldaferri F, Sans M, Vetrano S, Correale C, Arena V, Pagano N, Rando G, Romeo F, Potenza AE, Repici A, Malesci A, Danese S. (2009). The role of MAPK in governing lymphocyte adhesion to and migration across the microvasculature in inflammatory bowel disease. European Journal of Immunology 39: 290–300. DOI 10.1002/eji.200838316. [Google Scholar] [CrossRef]
Scher JU, Littman DR, Abramson SB. (2016). Microbiome in inflammatory arthritis and human rheumatic diseases. Arthritis & Rheumatology 68: 35–45. DOI 10.1002/art.39259. [Google Scholar] [CrossRef]
Schinzari F, Armuzzi A, De Pascalis B, Mores N, Tesauro M, Melina D, Cardillo C. (2008). Tumor necrosis factor-α antagonism improves endothelial dysfunction in patients with Crohn’s disease. Clinical Pharmacology & Therapeutics 83: 70–76. DOI 10.1038/sj.clpt.6100229. [Google Scholar] [CrossRef]
Schuermann GM, Aber-Bishop AE, Facer P, Lee JC, Rampton DS, Dore CJ, Polak JM. (1993). Altered expression of cell adhesion molecules in uninvolved gut in inflammatory bowel disease. Clinical & Experimental Immunology 94: 341–347. DOI 10.1111/j.1365-2249.1993.tb03455.x. [Google Scholar] [CrossRef]
Seldin MM, Meng Y, Qi H, Zhu WF, Wang Z, Hazen SL, Lusis AJ, Shih DM. (2016). Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. Journal of the American Heart Association 5: e002767. DOI 10.1161/JAHA.115.002767. [Google Scholar] [CrossRef]
Sen U, Mishra PK, Tyagi N, Tyagi SC. (2010). Homocysteine to hydrogen sulfide or hypertension. Cell Biochemistry and Biophysics 57: 49–58. DOI 10.1007/s12013-010-9079-y. [Google Scholar] [CrossRef]
Serviddio G, Bellanti F, Vendemiale G, Altomare E. (2011). Mitochondrial dysfunction in nonalcoholic steatohepatitis. Expert Review of Gastroenterology & Hepatology 5: 233–244. DOI 10.1586/egh.11.11. [Google Scholar] [CrossRef]
Shearn CT, Fennimore B, Orlicky DJ, Gao YR, Saba LM, Battista KD, Aivazidis S, Assiri M, Harris PS, Michel C, Merrill GF, Schmidt EE, Colgan SP, Petersen DR. (2019). Cholestatic liver disease results increased production of reactive aldehydes and an atypical periportal hepatic antioxidant response. Free Radical Biology and Medicine 143: 101–114. DOI 10.1016/j.freeradbiomed.2019.07.036. [Google Scholar] [CrossRef]
Shearn CT, Orlicky DJ, Petersen DR. (2018). Dysregulation of antioxidant responses in patients diagnosed with concomitant Primary Sclerosing Cholangitis/Inflammatory Bowel Disease. Experimental and Molecular Pathology 104: 1–8. DOI 10.1016/j.yexmp.2017.11.012. [Google Scholar] [CrossRef]
Shemer A, Erny D, Jung S, Prinz M. (2015). Microglia plasticity during health and disease: An immunological perspective. Trends in Immunology 36: 614–624. DOI 10.1016/j.it.2015.08.003. [Google Scholar] [CrossRef]
Sheth T, Pitchumoni CS, Das KM. (2015). Management of musculoskeletal manifestations in inflammatory bowel disease. Gastroenterology Research and Practice 2015: 387891. DOI 10.1155/2015/387891. [Google Scholar] [CrossRef]
Siener R, Bangen U, Sidhu H, Honow R, von Unruh G, Hesse A. (2013). The role of Oxalobacter formigenes colonization in calcium oxalate stone disease. Kidney International 83: 1144–1149. DOI 10.1038/ki.2013.104. [Google Scholar] [CrossRef]
Silva J, Brito BS, Silva INN, Nóbrega VG, Silva MCSM, Gomes HDN, Fortes FM, Pimentel AM, Mota J, Almeida N, Surlo VC, Lyra A, Rocha R, Santana GO. (2019). Frequency of hepatobiliary manifestations and concomitant liver disease in inflammatory bowel disease patients. BioMed Research International 2019: 7604939. DOI 10.1155/2019/7604939. [Google Scholar] [CrossRef]
Silverman MD, Tumuluri RJ, Davis M, Lopez G, Rosenbaum JT, Lelkes PI. (2002). Homocysteine upregulates vascular cell adhesion molecule-1 expression in cultured human aortic endothelial cells and enhances monocyte adhesion. Arteriosclerosis, Thrombosis, and Vascular Biology 22: 587–592. DOI 10.1161/01.ATV.0000014221.30108.08. [Google Scholar] [CrossRef]
Singer II, Kawka DW, Scott S, Weidner JR, Mumford RA, Riehl TE, Stenson WF. (1996). Expression of inducible nitric oxide synthase and nitrotyrosine in colonic epithelium in inflammatory bowel disease. Gastroenterology 111: 871–885. DOI 10.1016/S0016-5085(96)70055-0. [Google Scholar] [CrossRef]
Singh S, Kumar N, Loftus EVJr, Kane SV. (2013). Neurologic complications in patients with inflammatory bowel disease: Increasing relevance in the era of biologics. Inflammatory Bowel Disease 19: 864–872. DOI 10.1002/ibd.23011. [Google Scholar] [CrossRef]
Siva S, Barrack ER, Reddy GPV, Thamilselvan V, Thamilselvan S, Menon M, Bhandari M. (2009). A critical analysis of the role of gut Oxalobacter formigenes in oxalate stone disease. BJU International 103: 18–21. DOI 10.1111/j.1464-410X.2008.08122.x. [Google Scholar] [CrossRef]
Speca S, Dubuquoy L. (2017). Chronic bowel inflammation and inflammatory joint disease: Pathophysiology. Joint Bone Spine 84: 417–420. DOI 10.1016/j.jbspin.2016.12.016. [Google Scholar] [CrossRef]
Stadnicki A. (2012). Involvement of coagulation and hemostasis in inflammatory bowel diseases. Current Vascular Pharmacology 10: 659–669. DOI 10.2174/157016112801784495. [Google Scholar] [CrossRef]
Stewart CS, Duncan SH, Cave DR. (2004). Oxalobacter formigenes and its role in oxalate metabolism in the human gut. FEMS Microbiology Letters 230: 1–7. DOI 10.1016/S0378-1097(03)00864-4. [Google Scholar] [CrossRef]
Suh HY, Lee WJ, Na SY. (2019). Dermatologic manifestations in inflammatory bowel disease. Korean J Gastroenterol 73: 285–293. [Google Scholar]
Sunny NE, Parks EJ, Browning JD, Burgess SC. (2011). Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metabolism 14: 804–810. DOI 10.1016/j.cmet.2011.11.004. [Google Scholar] [CrossRef]
Szeto CC, McIntyre CW, Li PK (2018a). Circulating bacterial fragments as cardiovascular risk factors in CKD. Journal of the American Society of Nephrology 29: 1601–1608. DOI 10.1681/ASN.2018010068. [Google Scholar] [CrossRef]
Szeto W, van der Bent A, Petty CR, Reich J, Farraye F, Fishman LN (2018b). Use of social media for health-related tasks by adolescents with inflammatory bowel disease: A step in the pathway of transition. Inflammatory Bowel Diseases 24: 1114–1122. DOI 10.1093/ibd/izy021. [Google Scholar] [CrossRef]
Takahashi K, Nishida A, Fujimoto T, Fujii M, Shioya M, Imaeda H, Inatomi O, Bamba S, Sugimoto M, Andoh A. (2016). Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn’s Disease. Digestion 93: 59–65. [Google Scholar]
Takemura T, Okada M, Yagi K, Kuwajima H, Yanagida H. (2002). An adolescent with IgA nephropathy and Crohn disease: Pathogenetic implications. Pediatric Nephrology 17: 863–866. DOI 10.1007/s00467-002-0943-x. [Google Scholar] [CrossRef]
Taleban S, Li D, Targan SR, Ippoliti A, Brant SR, Cho JH, Duerr RH, Rioux JD, Silverberg MS, Vasiliauskas EA, Rotter JI, Haritunians T, Shih DQ, Dubinsky M, Melmed GY, McGovern DPB. (2016). Ocular manifestations in inflammatory bowel disease are associated with other extra-intestinal manifestations, gender, and genes implicated in other immune-related traits. Journal of Crohn’s and Colitis 10: 43–49. DOI 10.1093/ecco-jcc/jjv178. [Google Scholar] [CrossRef]
Tang WH, Hazen SL. (2014). The contributory role of gut microbiota in cardiovascular disease. Journal of Clinical Investigation 124: 4204–4211. DOI 10.1172/JCI72331. [Google Scholar] [CrossRef]
Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, Balish E, Hammer RE. (1994). The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. Journal of Experimental Medicine 180: 2359–2364. DOI 10.1084/jem.180.6.2359. [Google Scholar] [CrossRef]
Timani S, Mutasim DF. (2008). Skin manifestations of inflammatory bowel disease. Clinics in Dermatology 26: 265–273. DOI 10.1016/j.clindermatol.2007.10.018. [Google Scholar] [CrossRef]
Tito RY, Cypers H, Joossens M, Varkas GB, Van Praet L, Glorieus E, Van den Bosch F, De Vos M, Raes J, Elewaut D. (2017). Brief report: Dialister as a microbial marker of disease activity in spondyloarthritis. Arthritis & Rheumatology 69: 114–121. DOI 10.1002/art.39802. [Google Scholar] [CrossRef]
Tsaitas C, Semertzidou A, Sinakos E. (2014). Update on inflammatory bowel disease in patients with primary sclerosing cholangitis. World Journal of Hepatology 6: 178–187. DOI 10.4254/wjh.v6.i4.178. [Google Scholar] [CrossRef]
Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. (2005). Mechanisms of homocysteine-induced oxidative stress. American Journal of Physiology-Heart and Circulatory Physiology 289: H2649–H2656. DOI 10.1152/ajpheart.00548.2005. [Google Scholar] [CrossRef]
van Assche G, Dignass A, Bokemeyer B, Danese S, Gionchetti P, Moser G, Beaugerie L, Gomollón F, Häuser W, Herrlinger K, Oldenburg B, Panes J, Portela F, Rogler G, Stein JC, Tilg H, Travis S, Lindsay JO. (2013). Second European evidence-based consensus on the diagnosis and management of ulcerative colitis part 3: Special situations. Journal of Crohn’s and Colitis 7: 1–33. DOI 10.1016/j.crohns.2012.09.005. [Google Scholar] [CrossRef]
van Hogezand RA, Hamdy NA. (2006). Skeletal morbidity in inflammatory bowel disease. Scandinavian Journal of Gastroenterology Supplement 243: 59–64. DOI 10.1080/00365520600664276. [Google Scholar] [CrossRef]
van Staa TP. (2006). The pathogenesis, epidemiology and management of glucocorticoid-induced osteoporosis. Calcified Tissue International 79: 129–137. DOI 10.1007/s00223-006-0019-1. [Google Scholar] [CrossRef]
van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. (2005). Use of oral corticosteroids and risk of fractures. Journal of Bone and Mineral Research 20: 1487–1494, discussion 1486. [Google Scholar]
Vavricka SR, Brun L, Ballabeni P, Pittet V, Vavricka BMP, Zeitz J, Rogler G, Schoepfer AM. (2011). Frequency and risk factors for extraintestinal manifestations in the Swiss inflammatory bowel disease cohort. American Journal of Gastroenterology 106: 110–119. DOI 10.1038/ajg.2010.343. [Google Scholar] [CrossRef]
Vavricka SR, Schoepfer A, Scharl M, Lakatos PL, Navarini A, Rogler G. (2015). Extraintestinal manifestations of inflammatory bowel disease. Inflammatory Bowel Disease 21: 1982–1992. DOI 10.1097/MIB.0000000000000392. [Google Scholar] [CrossRef]
Venkatesh PG, Navaneethan U, Shen B. (2011). Hepatobiliary disorders and complications of inflammatory bowel disease. Journal of Digestive Diseases 12: 245–256. DOI 10.1111/j.1751-2980.2011.00511.x. [Google Scholar] [CrossRef]
Vernon G, Baranova A, Younossi ZM. (2011). Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Alimentary Pharmacology & Therapeutics 34: 274–285. DOI 10.1111/j.1365-2036.2011.04724.x. [Google Scholar] [CrossRef]
Villaran RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RDM, Argüelles S, Delgado-Cortés MDJ, Sobrino V, Van Rooijen N, Venero JL, Herrera AJ, Cano J, Machado A. (2010). Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: Potential risk factor in Parkinson’s disease. Journal of Neurochemistry 114: 1687–1700. DOI 10.1111/j.1471-4159.2010.06879.x. [Google Scholar] [CrossRef]
Villumsen M, Aznar S, Pakkenberg B, Jess T, Brudek T. (2019). Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977-2014. Gut 68: 18–24. DOI 10.1136/gutjnl-2017-315666. [Google Scholar] [CrossRef]
Walker JR, Ediger JP, Graff LA, Greenfeld JM, Clara I, Lix L, Rawsthorne P, Miller N, Rogala L, McPhail CM, Bernstein CN. (2008). The Manitoba IBD cohort study: A population-based study of the prevalence of lifetime and 12-month anxiety and mood disorders. American Journal of Gastroenterology 103: 1989–1997. DOI 10.1111/j.1572-0241.2008.01980.x. [Google Scholar] [CrossRef]
Wang G, Woo CW, Sung FL, Siow YL, Karmin O. (2002). Increased monocyte adhesion to aortic endothelium in rats with hyperhomocysteinemia: Role of chemokine and adhesion molecules. Arteriosclerosis, Thrombosis, and Vascular Biology 22: 1777–1783. DOI 10.1161/01.ATV.0000035404.18281.37. [Google Scholar] [CrossRef]
Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ, Hazen SL. (2015). Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163: 1585–1595. DOI 10.1016/j.cell.2015.11.055. [Google Scholar] [CrossRef]
Wang Z, Zhao Y. (2018). Gut microbiota derived metabolites in cardiovascular health and disease. Protein & Cell 9: 416–431. DOI 10.1007/s13238-018-0549-0. [Google Scholar] [CrossRef]
Wei W, Ding M, Zhou K, Xie H, Zhang M, Zhang C. (2017). Protective effects of wedelolactone on dextran sodium sulfate induced murine colitis partly through inhibiting the NLRP3 inflammasome activation via AMPK signaling. Biomedicine & Pharmacotherapy 94: 27–36. DOI 10.1016/j.biopha.2017.06.071. [Google Scholar] [CrossRef]
Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, Gonen A, Butler S, Witztum JL, Glass CK, Miller YI. (2010). Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via NF-kappaB and AP-1: Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circulation Research 107: 56–65. DOI 10.1161/CIRCRESAHA.110.218420. [Google Scholar] [CrossRef]
Worcester EM. (2002). Stones from bowel disease. Endocrinology and Metabolism Clinics of North America 31: 979–999. DOI 10.1016/S0889-8529(02)00035-X. [Google Scholar] [CrossRef]
Wu P, Jia F, Zhang B, Zhang P. (2017). Risk of cardiovascular disease in inflammatory bowel disease. Experimental and Therapeutic Medicine 13: 395–400. DOI 10.3892/etm.2016.3966. [Google Scholar] [CrossRef]
Yang L, Wang L, Wang X, Xian CJ, Lu H. (2016). A possible role of intestinal microbiota in the pathogenesis of ankylosing spondylitis. International Journal of Molecular Sciences 17: 12. [Google Scholar]
Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X, Zhao X, Li NN, Li S, Xue G, Cheng W, Li B, Li H, Lin W, Tian C, Zhao J, Han J, An D, Zhang Q, Wei H, Zheng M, Ma X, Li W, Chen X, Zhang Z, Zeng H, Ying S, Wu JX, Yang R, Liu D. (2019). Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metabolism 30: 1172. DOI 10.1016/j.cmet.2019.11.006. [Google Scholar] [CrossRef]
Zanoli L, Rastelli S, Inserra G, Castellino P. (2015). Arterial structure and function in inflammatory bowel disease. World Journal of Gastroenterology 21: 11304–11311. DOI 10.3748/wjg.v21.i40.11304. [Google Scholar] [CrossRef]
Zezos P, Kouklakis G, Saibil F. (2014). Inflammatory bowel disease and thromboembolism. World Journal of Gastroenterology 20: 13863–13878. DOI 10.3748/wjg.v20.i38.13863. [Google Scholar] [CrossRef]
Zhang Y, Ye P, Luo L, Bai Y, Xu R, Xiao W, Liu D, Wu H. (2014). Association between arterial stiffness and risk of coronary artery disease in a community-based population. Chinese Medical Journal 127: 3944–3947. [Google Scholar]
Zhao C, Ling Z, Newman MB, Bhatia A, Carvey PM. (2007). TNF-alpha knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiology of Disease 26: 36–46. DOI 10.1016/j.nbd.2006.11.012. [Google Scholar] [CrossRef]
Zhou W, Cao Q, Peng Y, Zhang QJ, Castrillon DH, DePinho RA, Liu ZP. (2009). FoxO4 inhibits NF-κB and protects mice against colonic injury and inflammation. Gastroenterology 137: 1403–1414. DOI 10.1053/j.gastro.2009.06.049. [Google Scholar] [CrossRef]
Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, Sartor RB, McIntyre TM, Silverstein RL, Tang WHW, DiDonato JA, Brown JM, Lusis AJ, Hazen SL. (2016). Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 165: 111–124. DOI 10.1016/j.cell.2016.02.011. [Google Scholar] [CrossRef]
Zieman SJ, Melenovsky V, Kass DA. (2005). Mechanisms, pathophysiology, and therapy of arterial stiffness. Arteriosclerosis, Thrombosis, and Vascular Biology 25: 932–943. DOI 10.1161/01.ATV.0000160548.78317.29. [Google Scholar] [CrossRef]
Zmora N, Levy M, Pevsner-Fishcer M, Elinav E. (2017). Inflammasomes and intestinal inflammation. Mucosal Immunology 10: 865–883. DOI 10.1038/mi.2017.19. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |