DOI:10.32604/biocell.2021.014453
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.014453 | www.techscience.com/journal/biocell |
| Article |
FBXW7 regulates epithelial barrier impairment in human bronchial epithelial cells in vitro by targeting apoptosis signal-regulating kinase1 via the p38 pathway
Department of Pediatrics, Shanghai Ninth People’s Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, 200011, China
*Address correspondence to: XiaoYing Zhang, songjr92006@126.com
#These authors contributed equally to this work
Received: 28 September 2020; Accepted: 21 December 2020
Abstract: Bronchial asthma is a common chronic inflammatory disease characterized by airway hyperresponsiveness (AHR), inflammatory cell infiltration, and airway remodeling. F-box/WD repeat-containing protein 7 (FBXW7), an E3 ubiquitin ligase, is required for various endothelial functions, such as cell migration, inflammation, and endothelial integrity. This study aimed to investigate the role of FBXW7 in lipopolysaccharide (LPS)-induced epithelial barrier impairment in bronchial epithelial cells in vitro. By using lentivirus-based technology, FBXW7 was overexpressed or silenced (24 h) in human bronchial epithelial (16HBE) cells, which were treated with LPS or not (24 h). Immunoprecipitation (IP) detection and Western blot analysis were used to evaluate the interaction of target proteins. Cell permeability was measured using transepithelial electrical resistance and FITC dextran flux (48 h). IL-1β, IL-18 and TNF-α in cell supernatants were measured using ELISA (48 h). The results showed that LPS stimulation suppressed FBXW7 expression in a time- and dose-dependent manner. LPS exposure decreased cell proliferation, elevated IL-1β, IL-18 and TNF-α, increased epithelial permeability, and p38 phosphorylation. These LPS-induced changes were partly compromised by FBXW7 overexpression. Similar to LPS stimulation, FBXW7 knockdown increased epithelial permeability and levels of inflammatory cytokines and p38 phosphorylation, which were, in part, blocked by apoptosis signal-regulating kinase (ASK) 1 knockdown or p38 pathway inhibition. IP and Western blot analysis showed that FBXW7 interacted with ASK1. ASK1 expression was inversely associated with FBXW7 expression. FBXW7 overexpression markedly enhanced ASK1 ubiquitination. These data revealed that FBXW7 counter against inflammation and protects epithelial barrier integrity in bronchial epithelial cells by promoting ubiquitination-mediated degradation of ASK1 via the p38 pathway.
Keywords: Bronchial epithelial cells; Inflammation; Lipopolysaccharide; Transepithelial electrical resistance; FITC dextran flux
Abbreviations
| AHR: | airway hyperresponsiveness |
| FBXW7: | F-box/WD repeat-containing protein 7 |
| LPS: | lipopolysaccharide |
| IP: | Immunoprecipitation |
| ASK: | apoptosis signal-regulating kinase |
| TJs: | tight junctions |
| AJs: | adherens junctions |
| ZO: | zonula occludens |
| MAP3K5: | mitogen-activated protein kinase kinase kinase 5 |
| IL-1β: | interleukin-1β |
| TNF-α: | tumor necrosis factor-α |
| JNK: | c-Jun N-terminal kinase |
Bronchial asthma is a common chronic inflammatory disease characterized by airway hyperresponsiveness (AHR), inflammatory cell infiltration, and airway remodeling (Michalik et al., 2018; Nanda and Wasan, 2020). Airway epithelium acts as a frontline defense against penetration of foreign substances and is implicated in regulating innate and adaptive immune responses in asthma (Mertens et al., 2017). Airway epithelial barrier dysfunction is critical for the onset and progression of asthma (Georas and Rezaee, 2014; Gon and Hashimoto, 2018). Apical junctional complexes play important roles in maintaining airway epithelial integrity and are composed of tight junctions (TJs) and adherens junctions (AJs) (Tatsuta et al., 2019). TJ proteins, such as zonula occludens (ZO) and claudin proteins, and AJ proteins, such as E-cadherin, have been demonstrated to be important components of the junctional complexes (Coopman and Djiane, 2016).
Airway inflammation is central to current asthma management. Lipopolysaccharide (LPS) may induce IL-8 or neutrophil inflammation in the airways of patients with severe asthma. A study has related LPS levels to airway neutrophils or IL-8 in the bronchoalveolar lavage fluid of asthmatic children (Hauk et al., 2008). Moreover, the bronchoalveolar lavage fluid LPS and genes related to the activation of LPS signaling were higher in corticosteroid-resistant asthma (Goleva et al., 2008). 16HBE cell line (originally a cystic fibrosis cell line) is usually used for in vitro models of bronchial asthma (Yin et al., 2019; Lan et al., 2020). This cell line is readily accessible and easy to manipulate and is widely used to analyze pathways related to barrier function in response to environmental agents (Vinhas et al., 2011; Heijink et al., 2012). However, such immortalized cell line is not “normal,” and they do not possess the underlying genetic or epigenetic features, which contribute to the disease phenotypes of asthma patients (Blume and Davies, 2013).
Apoptosis signal-regulating kinase (ASK) 1, namely, mitogen-activated protein kinase kinase kinase 5 (MAP3K5), belongs to the MAPKKK family, playing an essential role in a wide range of diseases, such as inflammatory diseases and cancer (Fujisawa, 2017). There is evidence that ASK1 is involved in AHR sensitivity and cytokine production in mice models with bronchial asthma stimulated by ovalbumin and may serve as a promising therapeutic target of asthma (Takada et al., 2013). ASK1 inhibition exerts a protective effect against hyperoxia-induced lung injury by suppressing the production of interleukin (IL)-1β and tumor necrosis factor (TNF)-α, recruitment of immune cells, and cell apoptosis (Fukumoto et al., 2016). ASK1 is involved in activating various signaling pathways governing cell apoptosis, growth, and differentiation, such as c-Jun N-terminal kinase (JNK) and p38 pathways (Cheon et al., 2018). P38 signaling pathway has been considered as a classical inflammatory pathway (Yeung et al., 2018). In addition, various pathways, such as NF-κB, HIF-1, and β-catenin, have been reported to be involved in the occurring and progression of bronchial asthma (Jia et al., 2019; Yuan et al., 2019; Sun et al., 2020). For instance, JAX2 can prevent bronchial asthma by inhibiting MAPK/NF-kB inflammatory signaling (Yuan et al., 2019). β-catenin pathway regulates asthma airway remodeling by modulating c-myc and cyclin D1 via the p38 MAPK-dependent pathway (Jia et al., 2019).
F-box/WD repeat-containing protein 7 (FBXW7) is a subunit of E3 ubiquitin ligase complex for recognizing substrate and has been established to be a tumor suppressor in the regulation of a wide range of oncoproteins responsible for ubiquitination and proteasome degradation (Yeh et al., 2018; Lan and Sun, 2019). FBXW7 is required for various endothelial functions, such as cell migration, inflammation, and endothelial integrity (Wang et al., 2013; Pronk et al., 2019). In a preliminary analysis, we found that FBXW7 was a predicted E3 ligase of ASK1 using UbiBrowser software. We postulate that FBXW7/ASK1 may be involved in the pathophysiology of bronchial asthma. 16HBE cell line (originally a cystic fibrosis cell line) is usually used for in vitro models of bronchial asthma (Yin et al., 2019; Lan et al., 2020). To test this postulation, acute lung injury in bronchial epithelial (16HBE) cells was caused by LPS. FBXW7 was overexpressed or interfered, to investigate its effect on cell proliferation, the release of inflammatory cytokines, and cell permeability and unravel the underlying molecular mechanisms.
We cultured 16HBE airway epithelial cells, which was purchased from the Cell Bank of the Chinese Academy of Science Shanghai, China, in DMEM medium (Hyclone, SH30243.01; Logan, UT, US) supplemented with 10% FBS (GIBCO, 16000e044; Carlsbad, CA, USA) and 1% P/S (Solarbio, P1400, Beijing, China) in an incubator (37°C, 5% CO2). The cells were treated with 0, 1.25, 2.5, or 5 µg/mL LPS (derived from Escherichia coli O55:B5, HY-D1056, MedChemExpress) for 0, 12, 24, or 48 h.
For FBXW7 overexpression (oeFBXW7), the coding sequence of FBXW7 (NM_033632.3) was synthesized and integrated into pLVX-Puro. The following primers were used: FBXW7-F: 5’ -CGGAATTCATGAATCAGGAACTGCTCTCTGTG-3’ (EcoRI); FBXW7-R: 5’-CGGGATCCTCACTTCATGTCCACATCAAAGTC-3’ (BamHI).
The FBXW7 interference (shFBXW7) sequences integrated into pLKO.1-puro for FBXW7 silencing were as follows: shFBXW7-1 (721-739), CCATGCAAAGTCTCAGAAT; shFBXW7-2 (1195-1213), GGTTTCATACACAGTCCAT; shFBXW7-3 (1669-1687), GCAGTCCGCTGTGTTCAAT.
When 16HBE cells grow to 60–70% confluence in 6-well plates, the cells were transfected with oeFBXW7 (MOI = 5, 5 µL) and empty plasmids (vector, MOI = 5, 5 µL), or shFBXW7-1, shFBXW7-2, shFBXW7-3 (MOI = 5, 5 µL) and shNC (MOI = 5, 5 µL) using Lipofectamine 2000 reagent (Invitrogen, CA, USA) according to the manufacturer’ instructions. After 24-h transfection, serum-free medium was replaced by the medium containing 10% FBS for 48-h culture.
Cell Counting Kit-8 (CCK-8) assay
CCK-8 assay was performed with a Cell Proliferation and Cytotoxicity Assay Kit (SAB, CP002; USA). Briefly, 100 mL of 16HBE cells suspension (2 × 103 cells) was added to each well of a 96-well plate. The cells were transfected with oeFBXW7 or empty plasmid (vector). After 24 h-culture, the cells were treated with LPS or vehicle. CCK-8 solution (100 µL) was added to each well for 1 h incubation, and then the absorbance at 450 nm, indicating cell ability, was measured using a microplate reader (E8051, Promega).
Quantitative real-time PCR (qRT-PCR)
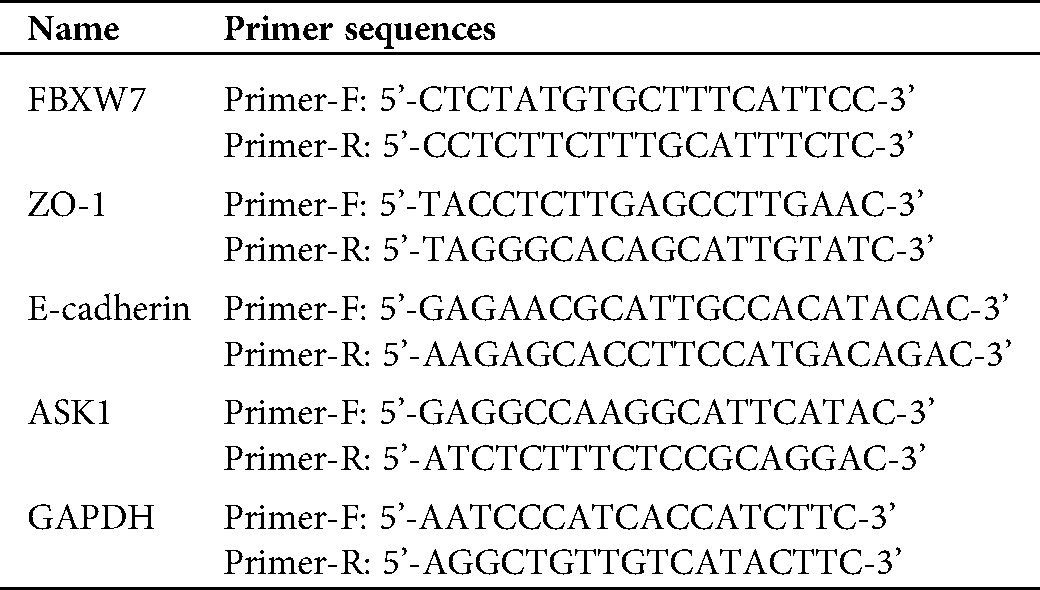
Briefly, RNA samples were reverse transcribed to cDNA using RevertAid First Strand cDNA Synthesis Kit (Fermentas, Hanover, MD, USA) and amplified with SYBR Green qPCR Master Mixes (#K0223, Thermo Fisher, Rockford, IL, USA) according to the standard protocol. The primer sequences, listed in Suppl. Tab. 1, were applied in the present study.
Enzyme-linked immunosorbent assay
Briefly, the cells were collected and centrifuged at 2000×g for 20 min following the required treatments. According to the manufacturer’s manual, IL-1β, IL-18, and TNF-α levels in supernatants were detected using ELISA Kits (Abcam, Cambridge, MA, USA).
Immunoprecipitation (IP) detection and Western blot analysis
IP and western blot analysis was conducted as previously described (Yang et al., 2017b; Zhang et al., 2019b). Protein A/G Plus-Agarose beads from Santa Cruz Biotechnology Inc (CA, USA) were used. Anti-FBXW7 (ab109617, Abcam) and anti-ASK1 (#8662, CST) antibodies were applied for IP detection. Anti-FBXW7 (ab105752), anti-ZO-1 (ab190085), anti-E-cadherin (ab40772), anti-p-p38 (ab195049), anti-p38 (ab31828) from Abcam, and anti-ASK1 (#3762), anti-GAPDH (#5174), and HRP-conjugated secondary antibodies from Cell Signaling Technology (MA, USA), were used for western blot analysis. The protein blots were estimated using a chemiluminescent imaging system (Tanon 5200, Shanghai, China).
Cell permeability was measured using two methods: transepithelial electrical resistance (TEER) and FITC dextran flux (Ryu et al., 2018). TEER was measured using a voltohmmeter (MillicellERS-2, Millipore). The resistance value was calculated as follows: TEER [Ω•cm2] = (R1 – R2) [Ω] × Effective membrane area [cm2], followed by calculation of electric resistance per unit area (R1: experimental group; R2: blank group). For dextran flux measurement, 10 kD FITC-conjugated dextran (sc-263323, Santa Cruz) was placed in the upper chamber of the transwell and incubated for 5 min, and then the supernatants from the lower chamber were collected to measure the base value. Later, the cells were supplemented with culture medium and incubated for 2 h. The supernatants from the lower chamber were collected to detect FITC fluorescence intensity using a microplate reader (E8051, Promega). The permeability (concentrations) was calculated according to the standard curve.
GraphPad Prism 7.0 software (San Diego, CA, USA) was used for all statistical analysis. Each experiment was performed in triplicate. Data were presented as mean value ± SD. One-way analysis of variance (ANOVA) with Tukey’s post hoc tests was applied for comparison of mean values between groups. p-value < 0.05 was considered statistically significant.
FBXW7 was lowly expressed in LPS stimulation-caused acute lung injury in human bronchial epithelial cells
Q-PCR and western blot showed that FBXW7 was lowly expressed in peripheral blood of patients with asthma (Figs. 1A, 1B). In order to investigate the impact of LPS on FBXW7 expression in 16HBE cells, the cells were stimulated by LPS at different concentrations (0, 1.25, 2.5, or 5 µg/mL) for different time durations (0, 12, 24, or 48 h). Expression of mRNA and protein of FBXW7 were remarkably decreased in response to the exposure of 2.5 or 5 µg/mL LPS for 24 h (p < 0.05, Figs. 1C–1D). Stimulation of 2.5 µg/mL LPS for 12, 24, or 48 h resulted in significant decreases in mRNA and protein of FBXW7 (Figs. 1E, 1F). These observations suggest that LPS stimulation inhibits FBXW7 expression in 16HBE cells in a dose- and time-dependent manner. Treatment of 2.5 µg/mL LPS for 24 h was selected to be used in further analysis.
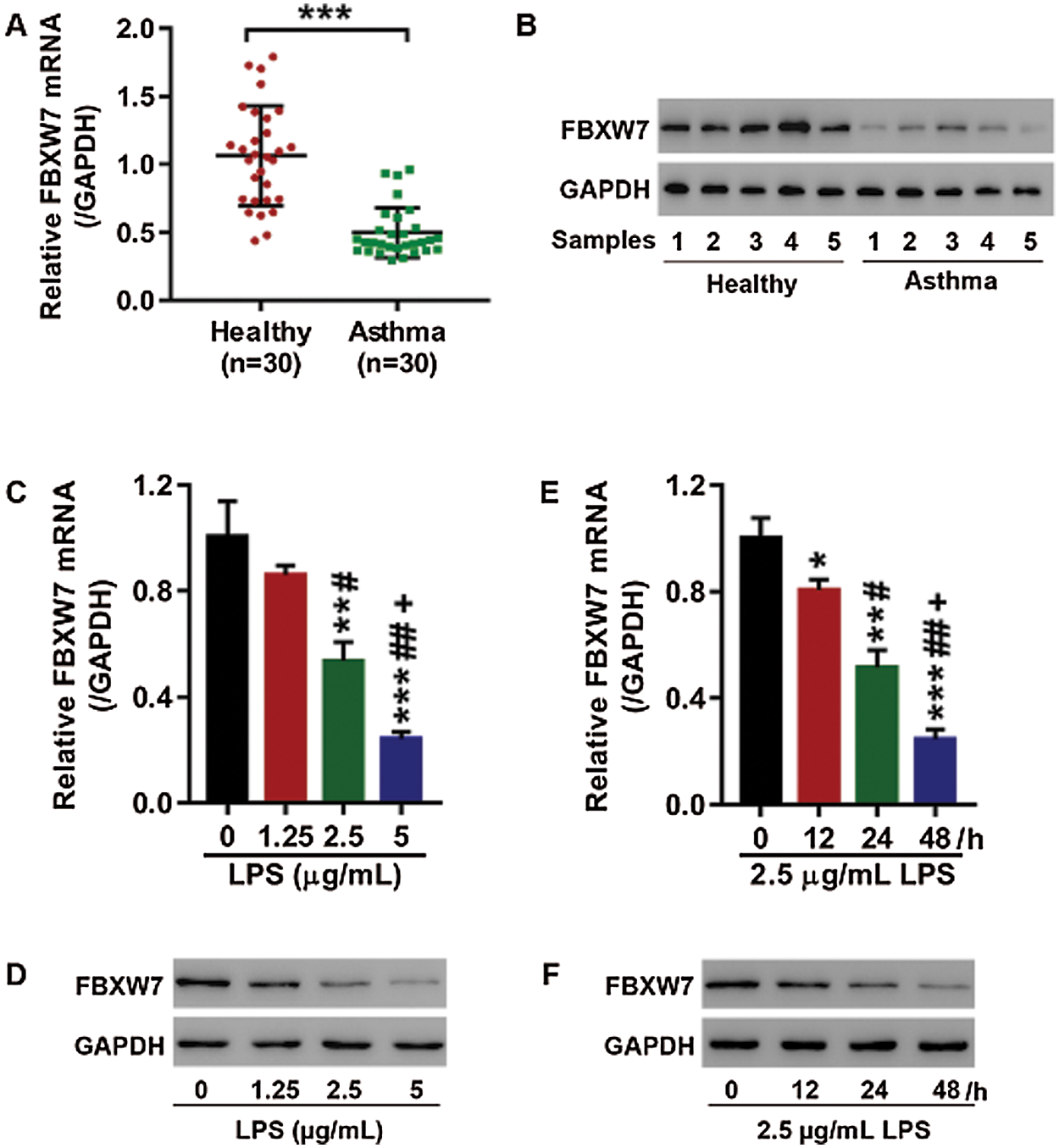
Figure 1: LPS suppresses FBXW7 expression in16HBEcells.
Overexpression of FBXW7 reversed LPS stimulation-caused cell proliferation inhibition, release of inflammatory cytokines and increased cell permeability
16HBE cells were transfected with oeFBXW7 to overexpress FBXW7 or empty vector prior to LPS stimulation (2.5 µg/mL, 24 h). As shown in Figs. 2A, 2B, FBXW7 was successfully overexpressed at mRNA and protein level in16HBE cells (p < 0.001). LPS exposure exerted a suppressive effect on cell proliferation (p < 0.01), which was partly restored by FBXW7 overexpression (p < 0.001, Fig. 2C). IL-1β, IL-18, and TNF-αwere obviously elevated in supernatants of the cells on exposure to LPS stimulation (p < 0.001, Figs. 2D–2F). And the LPS-induced release of IL-1β, IL-18, and TNF-α was partly compromised by FBXW7 overexpression (p < 0.01, Figs. 2D–2F). Moreover, to detect the effects of LPS and FBXW7 overexpression on cell permeability, TEER, FITC dextran flux, and expression of ZO-1 and E-cadherin were examined. LPS stimulation led to a precipitous decrease in TEER (p < 0.001), a remarkable increase in FITC dextran flux (p < 0.001), and obvious up-regulation of ZO-1 and E-cadherin at mRNA (p < 0.001) and protein level, which were all significantly suppressed by overexpressing FBXW7 (p < 0.01, Figs. 2G–2J). FBXW7 up-regulation alleviated the p38 phosphorylation stimulated by LPS as well (Fig. 2J). These results show that FBXW7 overexpression significantly reverses the impacts of LPS stimulation on cell proliferation, the release of inflammatory factors, cell permeability, and p38 pathway activation in 16HBE cells.
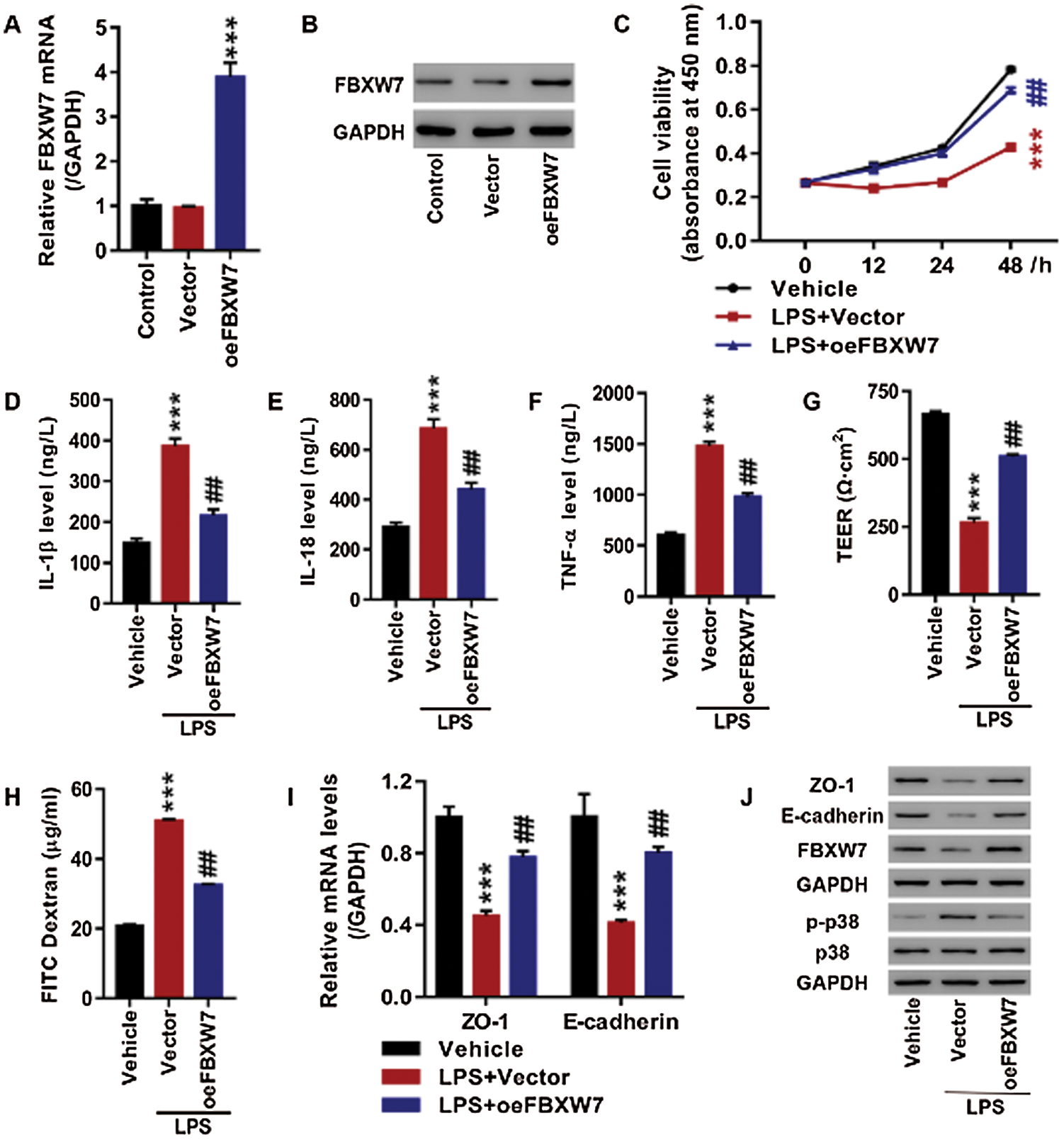
Figure 2: Up-regulation of FBXW7 significantly reverses the LPS-induced cell proliferation inhibition, release of inflammatory cytokines and increased cell permeability in 16HBE cells.
The interaction between FBXW7 and ASK1
FBXW7 was predicted to be an E3 ligase of ASK1 (MAP3K5) using UbiBrowser software. We tested whether ASK1 expression was regulated by FBXW7. 16HBE cells were transfected with oeFBXW7 or shFBXW7 to overexpress or silence FBXW7. FBXW7 overexpression resulted in a down-regulation of ASK1 protein, while FBXW7 knockdown caused an up-regulation ofASK1 protein (Fig. 3B). In sharp contrast, ASK1 mRNA expression was little affected by overexpressing or silencing FBXW7 (Fig. 3A). Besides, Western blot and IP analysis found interactions between FBXW7 and ASK1 (Fig. 3C). Furthermore, the ubiquitination of ASK is little after treatment of LPS (2.5 µg/mL), while FBXW7 overexpression dramatically promoted ASK1 ubiquitination in the LPS (2.5 µg/mL)-stimulated 16HBE cells co-treated with proteasome inhibitor MG132 or not (Fig. 3D). It implies thatFBXW7 interacts with ASK1 and affects ASK1 expression through modulating ubiquitination-mediated degradation of ASK1.
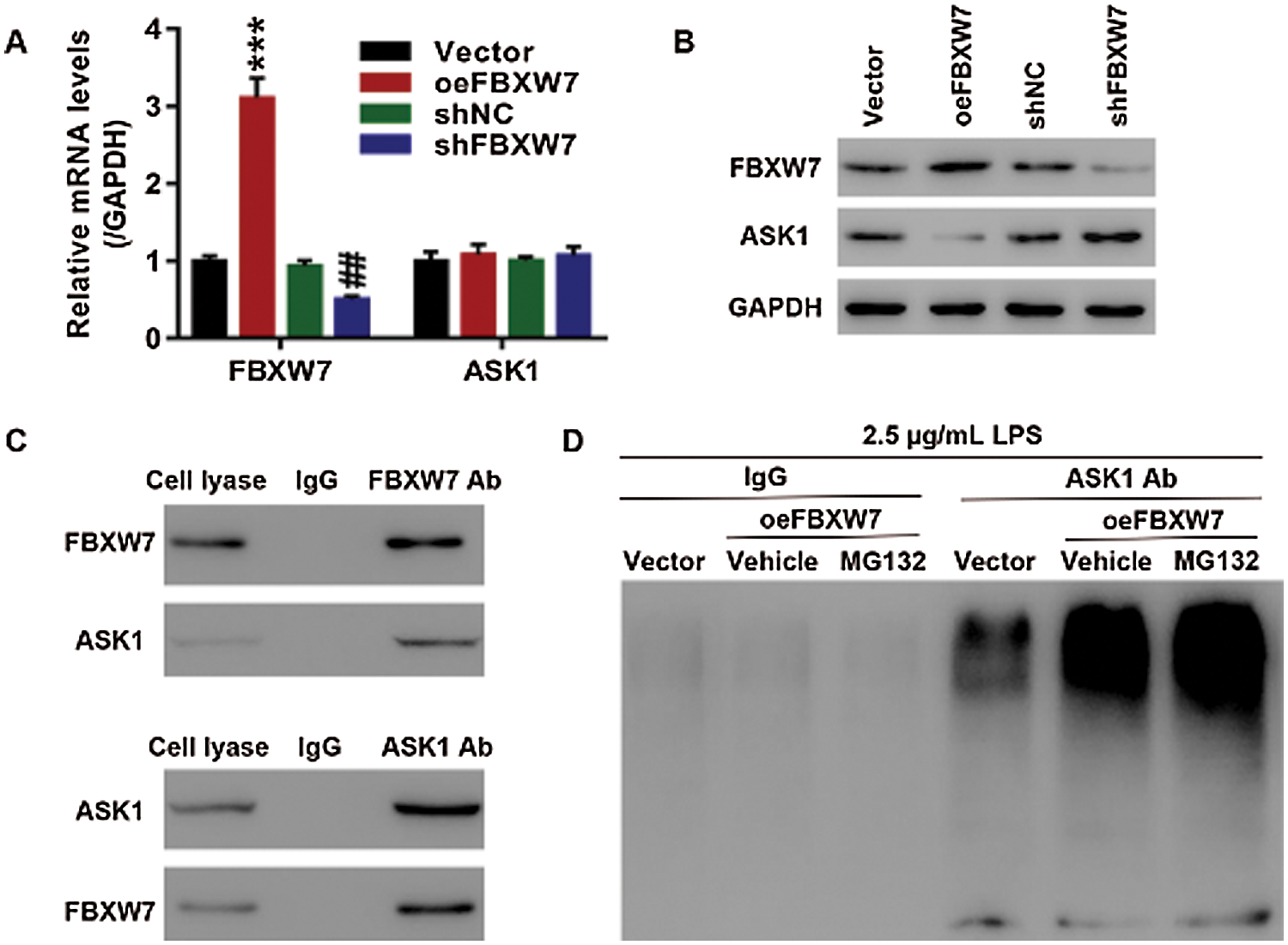
Figure 3: The interaction between FBXW7 and ASK1 in 16HBE cells. (A, B) FBXW7 expression is negatively associated with ASK1 expression at mRNA (A) and protein (B) level, ***p < 0.001 vs. vector; ##p < 0.01 vs. shNC. (C) Results of immunoprecipitation and western blot show that FBXW7 interacts with ASK1. (D) FBXW7 overexpression, in combination with LPS, enhances ASK1 ubiquitination. ASK1 ubiquitination is detected by immunoprecipitation and Western blot. The cells are transfected with oeFBXW7 or shFBXW7 to overexpress or silence FBXW7 expression. The cells transfected with oeFBXW7 or vector are then treated with 2.5 µg/mL LPS or 10 µmol/L MG132, or in combination, for 24 h.
FBXW7 regulated cell permeability in 16HBE cells probably by targeting ASK1 via p38 pathway
We further explored the role of ASK1 and p38 pathways in FBXW7 regulating the release of inflammatory factors and cell permeability of 16HBE cells. As depicted in Figs. 4A, 4B, FBXW7 was successfully silenced at mRNA and protein level in the 16HBE cells transfected with shFBXW7-1, shFBXW7-2, or shFBXW7-3. FBXW7 silencing markedly elevated IL-1β, IL-18, and TNF-α levels in the supernatants (p < 0.001, Figs. 4C–4E), decreased TEER (p < 0.001, Fig. 4F), increased FITC dextran flux (p < 0.001, Fig. 4G), down-regulated ZO-1 and E-cadherin at mRNA and protein level (p < 0.001, Figs. 4H, 4I), and up-regulated ASK1 and p-p38 protein (Fig. 4I), which were similar to the LPS-induced effects described above. Furthermore, in order to determine whether ASK1 and p38 pathways mediate the FBXW7 silencing-induced effects on inflammation and cell permeability, the shFBXW7-transfected cells were transfected with shASK1 to down-regulate ASK1 or treated with SB203580 to deactivate the p38 pathway. Either ASK1 knockdown or treatment of SB203580 attenuated the effects of FBXW7 knockdown on the release of inflammatory factors and cell permeability (Figs. 4C–4I). These results suggest that FBXW7 silencing may up-regulate ASK1 and activate the p38 pathway, thereby enhancing the release of IL-1β, IL-18, and TNF-α and increasing cell permeability in 16HBE cells.
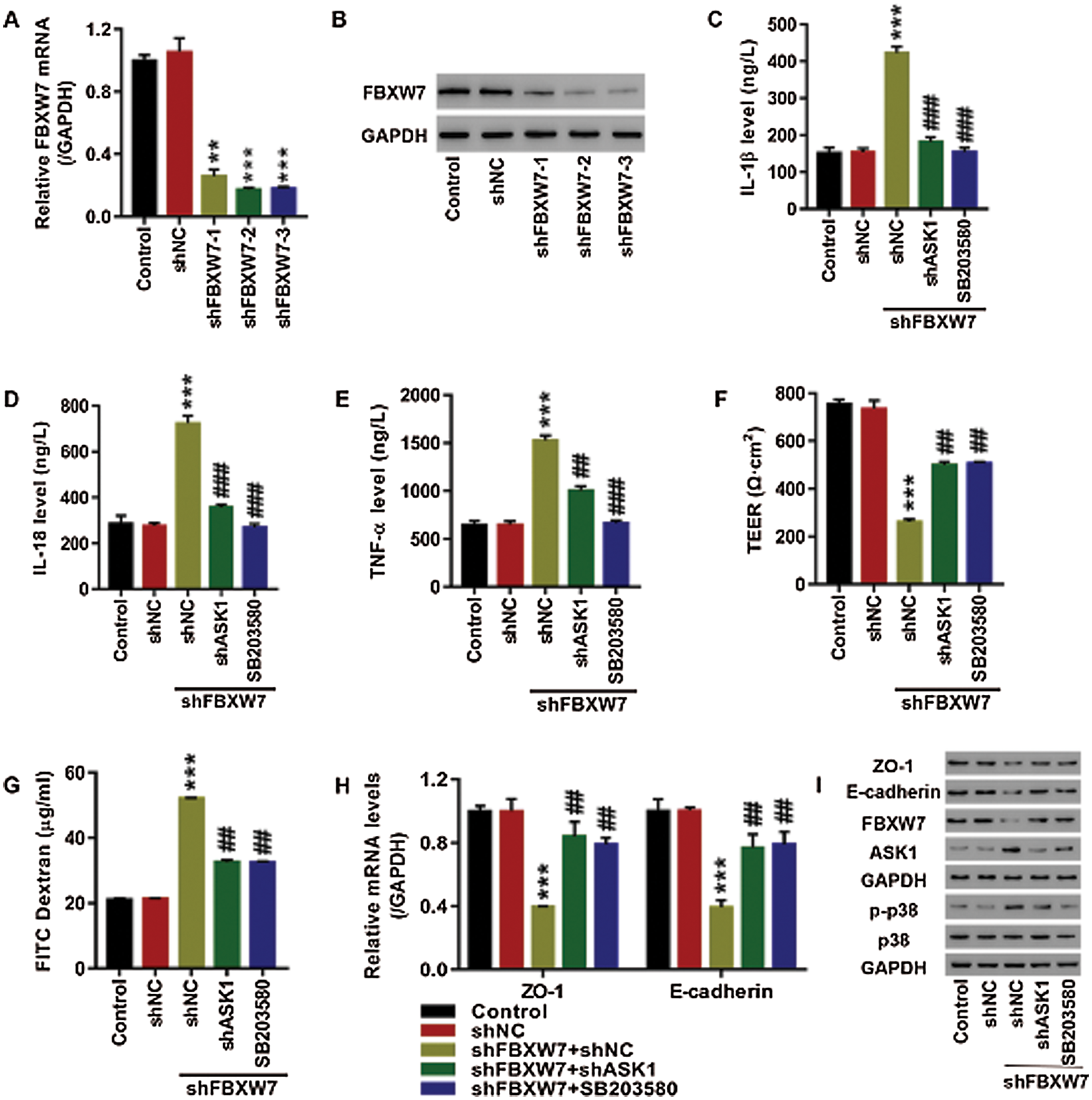
Figure 4: FBXW7 regulated cell permeability in 16HBE cells, probably by targeting ASK1 via the p38 pathway.
Previous studies have demonstrated the key involvement of FBXW7 in cancers and metabolism, suggesting the clinical potential of anticancer therapies against FBXW7 (Cao et al., 2016; Shimizu et al., 2018). For example, through promoting epithelial–mesenchymal transition, FBXW7 acts as a driver of uterine carcinosarcoma (Cuevas et al., 2019). It also reported that FBXW7-mediated STAT2 stability regulation plays an essential role in cell proliferation and cancer growth of melanoma (Lee et al., 2020). Besides, it has been established that FBXW7 is a potent regulator of angiogenesis (Yang et al., 2017a; Shao et al., 2019). Nevertheless, the implication of FBXW7 in the pathogenesis of bronchial asthma has been rarely studied. Previous studies have revealed that administration of LPS could establish the model of asthma, and exacerbate the inflammation, oxidative stress in an experimental model of asthma, as well as increased epithelial barrier permeability (Aggarwal et al., 2010; Camargo et al., 2017; Yu and Chen, 2018). Consistent with these, in the current study, 16HBE cells were treated with LPS to cause acute lung injury in vitro. We found that LPS stimulation exerted a suppressive effect on FBXW7 expression and cell proliferation and a stimulatory effect on the release of inflammatory cytokines (IL-1β, IL-18, and TNF-α) and cell permeability.
Some studies report that FBXW7 counters against inflammation (Balamurugan et al., 2013; Zhang et al., 2019a). Moreover, overexpression of FBXW7 has been found to abate the LPS-induced septic liver injury by inhibiting apoptosis and inflammation (Zhou and Xia, 2020). Nonetheless, a recent study holds a different viewpoint that FBXW7 promotes inflammation by acting as a novel E3 ubiquitin ligase for IκBα to degrade IκBα and activate NF-κB in inflammatory bowel disease (Meng et al., 2020). The current study showed that FBXW7 overexpression mitigated the LPS-induced release of IL-1β, IL-18, and TNF-α, whereas FBXW7 deficiency promoted the release of these inflammatory cytokines resembling LPS stimulation, supporting the protective role of FBXW7 against inflammation in bronchial epithelial cells. It can be speculated that FBXW7 has the potential to regulate inflammation positively and negatively through different mechanisms. These findings highlight the need for further studies on the regulatory effect of FBXW7 on inflammation and the underlying molecular mechanisms.
FBXW7 has long been established as a key regulator of endothelial barrier function (Wang et al., 2013). AJ protein E-cadherin and TJ protein ZO-1 are essential regulators of endothelial barrier functions and protect against tumor metastasis (Campbell et al., 2017). FBXW7 suppresses metastasis of various cancers, such as gastric cancer and lung cancer, by up-regulating E-cadherin expression (Huang et al., 2018; Zhang et al., 2018). In this study, either LPS stimulation or FBXW7 knockdown increased epithelial cell permeability, as evidenced by the observations of decreased TEER, increased FITC dextran flux, down-regulated ZO-1 and E-cadherin expression. Moreover, FBXW7 overexpression significantly decreased epithelial cell permeability, compromising the effect of LPS on epithelial cell permeability. Collectively, it can be concluded that FBXW7 expression protects epithelial barrier integrity and function. In accordance with our results, a recent study showed that loss of FBXW7 increases contractility and permeability of umbilical vein endothelial cells by accumulating RhoB GTPase (Pronk et al., 2019).
ASK1 is a key molecule in the ASK family that can be activated in response to a wide range of stressors and participates in the regulation of cellular processes, such as inflammation and cell apoptosis (Ryuno et al., 2017). Ubiquitination is crucial for ASK1 activation, and the E3 ligase FBXW5 has been revealed to be a critical activator of ASK1 ubiquitination (Bai et al., 2019). Our study suggests that FBXW7 overexpression promotes ASK1 ubiquitination, thus down-regulating ASK1 protein expression. Moreover, Western blot and IP analysis provide convincing evidence in support of FBXW7-ASK1 interaction. FBXW7 may be another activator of ASK1 ubiquitination. As far as we know, this is the first study uncovering associations of FBXW7 with ASK1. ASK1/2 signaling stimulates the inflammatory response, increases vascular endothelial permeability, promotes macrophages infiltration, and facilitates tumor metastasis (Tartey et al., 2018; Najafi et al., 2019). Consistently, the study found that interfering with ASK1 could partly abolish the FBXW7 silencing-induced elevations of inflammatory cytokines and increased cell permeability. It implies that FBXW7 may suppress inflammation and decrease bronchial epithelial permeability by down-regulating ASK1 via promoting ubiquitination-mediated ASK1 degradation.
P38 pathway is an important pathway regulating inflammatory response, and down-regulating the p38 pathway protects against inflammation (Zhou et al., 2017; Wu et al., 2018). Increasing evidence has pointed out that the p38 signaling pathway is involved in regulating asthmatic airway remodeling, the primary characteristic of asthma (Cao et al., 2018; Jia et al., 2019). P38 activation mediates the airway epithelial barrier dysfunction and asthma induced by toluene diisocyanate (Sulaiman et al., 2018). Similarly, in this study, inhibiting the p38 pathway alleviated inflammation and the increased epithelial permeability caused by FBXW7 silencing. Moreover, FBXW7 overexpression abrogated p38 pathway activation, indicating that FBXW7 may suppress inflammation and protects epithelial barrier integrity via the p38 pathway. ASK1 functions as an upstream regulator of the p38 pathway (Tesch et al., 2016). It can be speculated that FBXW7 may regulate inflammation and epithelial barrier integrity through regulating ASK1 via the p38 pathway.
However, there are also some limitations in the present research. One of the limitations is the lack of in vivo experiments in the current study. The next steps of our study are that need to be undertaken is looking at FBXW7 regulation on in vivo experiments. FBXW7 overexpression and KO mice would be highly informative to acute lung injury. Another is that only one cell line was used in our study. If possible, some other cell lines will be used to enrich our experimental content in the future. Although further investigations are needed, this study identifies a potential therapeutic approach for bronchial asthma.
In summary, this study demonstrates that FBXW7 suppresses the inflammatory response and decreases cell permeability in bronchial epithelial cells through boosting ubiquitination-mediated degradation of ASK1 via the p38 pathway. It also provides evidence that FBXW7 interacts with ASK1. This study not only stresses the relevance of FBXW7 as a potential therapeutic target in bronchial asthma but also mentions other pathologies for which FBXW7 might prove to be a potential target, which expands our understanding of the physiological contribution of FBXW7 to bronchial asthma, supporting the pursuit of FBXW7 as a potential therapeutic target against bronchial asthma.
Supplementary Table 1: The primer sequences
Availability of Data and Materials: All data generated or analyzed during this study are included in this published article.
Authors’ Contributions: XYZ conceived and designed the study. JRS, WL, and YD performed the experiments and collected and analyzed the data. JK wrote the manuscript. XYZ read and approved the final manuscript. All authors read and approved the final manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work is appropriately investigated and resolved.
Funding Statement: This study was funded by Fundamental research program funding of Ninth People’s Hospital Affiliated to Shanghai Jiaotong University School of Medicine (JYZZ069).
Conflicts of Interest: The authors declare that they have no competing interests.
Aggarwal NR, D’Alessio FR, Tsushima K, Files DC, Damarla M, Sidhaye VK, Fraig MM, Polotsky VY, King LS. (2010). Moderate oxygen augments lipopolysaccharide-induced lung injury in mice. American Journal of Physiology-Lung Cellular and Molecular Physiology 298: L371–L381. DOI 10.1152/ajplung.00308.2009. [Google Scholar] [CrossRef]
Bai L, Chen MM, Chen ZD, Zhang P, Tian S, Zhang Y, Zhu XY, Liu Y, She ZG, Ji YX, Li HL. (2019). F-box/WD repeat-containing protein 5 mediates the ubiquitination of apoptosis signal-regulating kinase 1 and exacerbates nonalcoholic steatohepatitis in mice. Hepatology 70: 1942–1957. DOI 10.1002/hep.30537. [Google Scholar] [CrossRef]
Balamurugan K, Sharan S, Klarmann KD, Zhang Y, Coppola V, Summers GH, Roger T, Morrison DK, Keller JR, Sterneck E. (2013). FBXW7α attenuates inflammatory signalling by downregulating C/EBPδ and its target gene Tlr4. Nature Communications 4: 637. DOI 10.1038/ncomms2677. [Google Scholar] [CrossRef]
Blume C, Davies DE. (2013). In vitro and ex vivo models of human asthma. European Journal of Pharmaceutics and Biopharmaceutics 84: 394–400. DOI 10.1016/j.ejpb.2012.12.014. [Google Scholar] [CrossRef]
Camargo LDN, Righetti RF, Aristóteles L, Dos Santos TM de Souza FCR, Fukuzaki S, Cruz MM, Alonso-Vale MIC, Saraiva-Romanholo BM, Prado CM, Martins MDA, Leick EA, Tibério I. (2018). Effects of anti-IL-17 on inflammation, remodeling, and oxidative stress in an experimental model of asthma exacerbated by LPS. Frontiers in Immunology 8: 193. DOI 10.3389/fimmu.2017.01835. [Google Scholar] [CrossRef]
Campbell HK, Maiers JL, DeMali KA. (2017). Interplay between tight junctions & adherens junctions. Experimental Cell Research 358: 39–44. DOI 10.1016/j.yexcr.2017.03.061. [Google Scholar] [CrossRef]
Cao J, Ge MH, Ling ZQ. (2016). Fbxw7 tumor suppressor: A vital regulator contributes to human tumorigenesis. Medicine 95: e2496. DOI 10.1097/MD.0000000000002496. [Google Scholar] [CrossRef]
Cao L, Liu F, Liu Y, Liu T, Wu J. (2018). TSLP promotes asthmatic airway remodeling via p38-STAT3 signaling pathway in human lung fibroblast. Experimental Lung Research 44: 288–301. DOI 10.1080/01902148.2018.1536175. [Google Scholar] [CrossRef]
Coopman P, Djiane A. (2016). Adherens Junction and E-Cadherin complex regulation by epithelial polarity. Cellular and Molecular Life Sciences 73: 3535–3553. DOI 10.1007/s00018-016-2260-8. [Google Scholar] [CrossRef]
Cuevas IC, Sahoo SS, Kumar A, Zhang H, Westcott J, Aguilar M, Cortez JD, Sullivan SA, Xing C, Hayes DN, Brekken RA, Bae-Jump VL, Castrillon DH. (2019). Fbxw7 is a driver of uterine carcinosarcoma by promoting epithelial-mesenchymal transition. Proceedings of the National Academy of Sciences of the United States of America 116: 25880–25890. DOI 10.1073/pnas.1911310116. [Google Scholar] [CrossRef]
Cheon SY, Kim EJ, Kim JM, Koo BN. (2018). Cell Type-specific mechanisms in the pathogenesis of ischemic stroke: the role of apoptosis signal-regulating kinase 1. Oxidative Medicine and Cellular Longevity 2018: 1–9. DOI 10.1155/2018/2596043. [Google Scholar] [CrossRef]
Fujisawa T. (2017). Therapeutic application of apoptosis signal-regulating kinase 1 inhibitors. Advances in Biological Regulation 66: 85–90. DOI 10.1016/j.jbior.2017.10.004. [Google Scholar] [CrossRef]
Fukumoto J, Cox R, Fukumoto I, Cho Y, Parthasarathy PT, Galam L, Lockey RF, Kolliputi N. (2016). Deletion of ASK1 Protects against Hyperoxia-Induced Acute Lung Injury. PLoS One 11: e0147652. DOI 10.1371/journal.pone.0147652. [Google Scholar] [CrossRef]
Georas SN, Rezaee F. (2014). Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. Journal of Allergy and Clinical Immunology 134: 509–520. DOI 10.1016/j.jaci.2014.05.049. [Google Scholar] [CrossRef]
Goleva E, Hauk PJ, Hall CF, Liu AH, Riches DWH, Martin RJ, Leung DYM. (2008). Corticosteroid-resistant asthma is associated with classical antimicrobial activation of airway macrophages. Journal of Allergy and Clinical Immunology 122: 550–559.e553. DOI 10.1016/j.jaci.2008.07.007. [Google Scholar] [CrossRef]
Gon Y, Hashimoto S. (2018). Role of airway epithelial barrier dysfunction in pathogenesis of asthma. Allergology International 67: 12–17. DOI 10.1016/j.alit.2017.08.011. [Google Scholar] [CrossRef]
Hauk PJ, Krawiec M, Murphy J, Boguniewicz J, Schiltz A, Goleva E, Liu AH, Leung DYM. (2008). Neutrophilic airway inflammation and association with bacterial lipopolysaccharide in children with asthma and wheezing. Pediatric Pulmonology 43: 916–923. DOI 10.1002/ppul.20880. [Google Scholar] [CrossRef]
Heijink IH, Brandenburg SM, Postma DS, van Oosterhout AJ. (2012). Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. European Respiratory Journal 39: 419–428. DOI 10.1183/09031936.00193810. [Google Scholar] [CrossRef]
Huang LY, Zhao J, Chen H, Wan L, Inuzuka H, Guo J, Fu X, Zhai Y, Lu Z, Wang X, Han ZG, Sun Y, Wei W. (2018). SCFFBW7-mediated degradation of Brg1 suppresses gastric cancer metastasis. Nature Communications 9: 143. DOI 10.1038/s41467-018-06038-y. [Google Scholar] [CrossRef]
Jia XX, Zhu TT, Huang Y, Zeng XX, Zhang H, Zhang WX. (2019). Wnt/β-catenin signaling pathway regulates asthma airway remodeling by influencing the expression of c-Myc and cyclin D1 via the p38 MAPK-dependent pathway. Experimental and Therapeutic Medicine 18: 3431–3438. [Google Scholar]
Lan H, Luo L, Chen Y, Wang M, Yu Z, Gong Y. (2020). MIF signaling blocking alleviates airway inflammation and airway epithelial barrier disruption in a HDM-induced asthma model. Cellular Immunology 347: 103965. DOI 10.1016/j.cellimm.2019.103965. [Google Scholar] [CrossRef]
Lan H, Sun Y. (2019). FBXW7 E3 ubiquitin ligase: Degrading, not degrading, or being degraded. Protein & Cell 10: 861–863. DOI 10.1007/s13238-019-0652-x. [Google Scholar] [CrossRef]
Lee CJ, An HJ, Kim SM, Yoo SM, Park J, Lee GE, Kim WY, Kim DJ, Kang HC, Lee JY, Lee HS, Cho SJ, Cho YY. (2020). FBXW7-mediated stability regulation of signal transducer and activator of transcription 2 in melanoma formation. Proceedings of the National Academy of Sciences of the United States of America 117: 584–594. DOI 10.1073/pnas.1909879116. [Google Scholar] [CrossRef]
Meng Q, Wu W, Pei T, Xue J, Xiao P, Sun L, Li L, Liang D. (2020). miRNA-129/FBW7/NF-κB, a novel regulatory pathway in inflammatory bowel disease. Molecular Therapy-Nucleic Acids 19: 731–740. [Google Scholar]
Mertens TCJ, Karmouty-Quintana H, Taube C, Hiemstra PS. (2017). Use of airway epithelial cell culture to unravel the pathogenesis and study treatment in obstructive airway diseases. Pulmonary Pharmacology & Therapy 45: 101–113. DOI 10.1016/j.pupt.2017.05.008. [Google Scholar] [CrossRef]
Michalik M, Wójcik-Pszczoła K, Paw M, Wnuk D, Koczurkiewicz P, Sanak M, Pekala E, Madeja Z. (2018). Fibroblast-to-myofibroblast transition in bronchial asthma. Cellular and Molecular Life Sciences 75: 3943–3961. DOI 10.1007/s00018-018-2899-4. [Google Scholar] [CrossRef]
Najafi M, Ahmadi A, Mortezaee K. (2019). Extracellular-signal-regulated kinase/mitogen-activated protein kinase signaling as a target for cancer therapy: An updated review. Cell Biology International 43: 1206–1222. DOI 10.1002/cbin.11187. [Google Scholar] [CrossRef]
Nanda A, Wasan AN. (2020). Asthma in adults. Medical Clinics 104: 95–108. [Google Scholar]
Pronk MCA, Majolée J, Loregger A, van Bezu JSM, Zelcer N, Hordijk PL, Kovačević I. (2019). FBXW7 regulates endothelial barrier function by suppression of the cholesterol synthesis pathway and prenylation of RhoB. Molecular Biology of the Cell 30: 607–621. DOI 10.1091/mbc.E18-04-0259. [Google Scholar] [CrossRef]
Ryu WI, Lee H, Bae HC, Jeon J, Ryu HJ, Kim J, Kim JH, Son JW, Kim JY, Imai Y, Yamanishi K, Jeong SH, Son SW. (2018). IL-33 down-regulates CLDN1 expression through the ERK/STAT3 pathway in keratinocytes. Journal of Dermatological Science 90: 313–322. DOI 10.1016/j.jdermsci.2018.02.017. [Google Scholar] [CrossRef]
Ryuno H, Naguro I, Kamiyama M. (2017). ASK family and cancer. Advances in Biological Regulation 66: 72–84. DOI 10.1016/j.jbior.2017.05.003. [Google Scholar] [CrossRef]
Shao J, Fan G, Yin X, Gu Y, Wang X, Xin Y, Yao Y. (2019). A novel transthyretin/STAT4/miR-223-3p/FBXW7 signaling pathway affects neovascularization in diabetic retinopathy. Molecular and Cellular Endocrinology 498: 110541. DOI 10.1016/j.mce.2019.110541. [Google Scholar] [CrossRef]
Shimizu K, Nihira NT, Inuzuka H, Wei W. (2018). Physiological functions of FBW7 in cancer and metabolism. Cellular Signalling 46: 15–22. DOI 10.1016/j.cellsig.2018.02.009. [Google Scholar] [CrossRef]
Sulaiman I, Tan K, Mohtarrudin N, Lim JCW, Stanslas J. (2018). Andrographolide prevented toluene diisocyanate-induced occupational asthma and aberrant airway E-cadherin distribution via p38 MAPK-dependent Nrf2 induction. Pulmonary Pharmacology & Therapeutics 53: 39–51. DOI 10.1016/j.pupt.2018.09.008. [Google Scholar] [CrossRef]
Sun L, Fu J, Lin SH, Sun JL Xia L, Lin CH, Liu L, Zhang C, Yang L, Xue P, Wang X, Huang S, Han X, Chen HL, Huang MS, Zhang X, Huang SK, Zhou Y. (2020). Particulate matter of 2.5 μm or less in diameter disturbs the balance of TH17/regulatory T cells by targeting glutamate oxaloacetate transaminase 1 and hypoxia-inducible factor 1α in an asthma model. Journal of Allergy and Clinical Immunology 145: 402–414. DOI 10.1016/j.jaci.2019.10.008. [Google Scholar] [CrossRef]
Takada E, Furuhata M, Nakae S, Ichijo H, Sudo K, Mizuguchi J. (2013). Requirement of apoptosis-inducing kinase 1 for the induction of bronchial asthma following stimulation with ovalbumin. International Archives of Allergy and Immunology 162: 104–114. DOI 10.1159/000353240. [Google Scholar] [CrossRef]
Tartey S, Gurung P, Dasari TK, Burton A, Kanneganti TD. (2018). ASK1/2 signaling promotes inflammation in a mouse model of neutrophilic dermatosis. Journal of Clinical Investigation 128: 2042–2047. DOI 10.1172/JCI98446. [Google Scholar] [CrossRef]
Tatsuta M, Kan-o K, Ishii Y, Yamamoto N, Ogawa T, Fukuyama S, Ogawa A, Fujita A, Nakanishi Y, Matsumoto K. (2019). Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: Protective role of cathelicidin LL-37. Respiratory Research 20: 899. DOI 10.1186/s12931-019-1226-4. [Google Scholar] [CrossRef]
Tesch GH, Ma FY, Nikolic-Paterson DJ. (2016). ASK1: A new therapeutic target for kidney disease. American Journal of Physiology-Renal Physiology 311: F373–F381. DOI 10.1152/ajprenal.00208.2016. [Google Scholar] [CrossRef]
Vinhas R, Cortes L, Cardoso I, Mendes VM, Manadas B, Todo-Bom A, Pires E, Veríssimo P. (2011). Pollen proteases compromise the airway epithelial barrier through degradation of transmembrane adhesion proteins and lung bioactive peptides. Allergy 66: 1088–1098. DOI 10.1111/j.1398-9995.2011.02598.x. [Google Scholar] [CrossRef]
Wang R, Wang Y, Liu N, Ren C, Jiang C, Zhang K, Yu S, Chen Y, Tang H, Deng Q, Fu C, Wang Y, Li R, Liu M, Pan W, Wang P. (2013). FBW7 regulates endothelial functions by targeting KLF2 for ubiquitination and degradation. Cell Research 23: 803–819. DOI 10.1038/cr.2013.42. [Google Scholar] [CrossRef]
Wu Y, Wang F, Fan L, Zhang W, Wang T, Du Y, Bai X. (2018). Baicalin alleviates atherosclerosis by relieving oxidative stress and inflammatory responses via inactivating the NF-κB and p38 MAPK signaling pathways. Biomedicine & Pharmacotherapy 97: 1673–1679. DOI 10.1016/j.biopha.2017.12.024. [Google Scholar] [CrossRef]
Yang M, Li CJ, Sun X, Guo Q, Xiao Y, Su T, Tu ML, Peng H, Lu Q, Liu Q, He HB, Jiang TJ, Lei MX, Wan M, Xu C, Luo XH (2017a). MiR-497∼195 cluster regulates angiogenesis during coupling with osteogenesis by maintaining endothelial Notch and HIF-1α activity. Nature Communications 8: 6382. DOI 10.1038/ncomms16003. [Google Scholar] [CrossRef]
Yang M, Yang Y, She S, Li S (2017b). Proteomic investigation of the effects of preimplantation factor on human embryo implantation. Molecular Medicine Reports 17: 3481–3488. [Google Scholar]
Yeh C, Bellon M, Nicot C. (2018). FBXW7: A critical tumor suppressor of human cancers. Molecular Cancer 17: 1852. DOI 10.1186/s12943-018-0857-2. [Google Scholar] [CrossRef]
Yeung YT, Aziz F, Guerrero-Castilla A, Arguelles S. (2018). Signaling pathways in inflammation and anti-inflammatory therapies. Current Pharmaceutical Design 24: 1449–1484. DOI 10.2174/1381612824666180327165604. [Google Scholar] [CrossRef]
Yin CY, Bai QF, Feng JX. (2019). MiR-216a-5p protects 16HBE cells from H2O2-induced oxidative stress through targeting HMGB1/NF-kB pathway. Biochemical and Biophysical Research Communications 508: 416–420. DOI 10.1016/j.bbrc.2018.11.060. [Google Scholar] [CrossRef]
Yu QL, Chen Z. (2018). Establishment of different experimental asthma models in mice. Experimental and Therapeutic Medicine 15: 2492–2498. [Google Scholar]
Yuan F, Liu R, Hu M, Rong X, Bai L, Xu L, Mao Y, Hasimu H, Sun Y, He J. (2019). JAX2, an ethanol extract of Hyssopus cuspidatus Boriss, can prevent bronchial asthma by inhibiting MAPK/NF-κB inflammatory signaling. Phytomedicine 57: 305–314. DOI 10.1016/j.phymed.2018.12.043. [Google Scholar] [CrossRef]
Zhang C, Chen F, Feng L, Shan Q, Zheng GH, Wang YJ, Lu J, Fan SH, Sun CH, Wu DM, Li MQ, Hu B, Wang QQ, Zhang ZF, Zheng YL (2019a). FBXW7 suppresses HMGB1-mediated innate immune signaling to attenuate hepatic inflammation and insulin resistance in a mouse model of nonalcoholic fatty liver disease. Molecular Medicine 25: 174. DOI 10.1186/s10020-019-0099-9. [Google Scholar] [CrossRef]
Zhang M, Wang X, Yao J, Qiu Z (2019b). Long non-coding RNA NEAT1 inhibits oxidative stress-induced vascular endothelial cell injury by activating the miR-181d-5p/CDKN3 axis. Artificial Cells 47: 3129–3137. [Google Scholar]
Zhang Y, Zhang X, Ye M, Jing P, Xiong J, Han Z, Kong J, Li M, Lai X, Chang N, Zhang J, Zhang J. (2018). FBW7 loss promotes epithelial-to-mesenchymal transition in non-small cell lung cancer through the stabilization of Snail protein. Cancer Letters 419: 75–83. DOI 10.1016/j.canlet.2018.01.047. [Google Scholar] [CrossRef]
Zhou P, Lu S, Luo Y, Wang S, Yang K, Zhai Y, Sun G, Sun X. (2017). Attenuation of TNF-α-induced inflammatory injury in endothelial cells by ginsenoside Rb1 via inhibiting NF-κB, JNK and p38 signaling pathways. Frontiers in Pharmacology 8: 26346. DOI 10.3389/fphar.2017.00464. [Google Scholar] [CrossRef]
Zhou YP, Xia Q. (2020). Inhibition of miR-103a-3p suppresses lipopolysaccharide-induced sepsis and liver injury by regulating FBXW7 expression. Cell Biology International 44: 1798–1810. DOI 10.1002/cbin.11372. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |