DOI:10.32604/biocell.2021.013236
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.013236 | www.techscience.com/journal/biocell |
| Article |
Upregulation of miR-143-3p attenuates oxidative stress-mediated cell ferroptosis in cardiomyocytes with atrial fibrillation by degrading glutamic-oxaloacetic transaminase 1
1Department of Functional Examination, The Sixth Affiliated Hospital of Xinjiang Medical University, Urumqi, 830000, China
2Health Care Third Department, Xinjiang Military Region General Hospital, Urumqi, 830000, China
3Department of Emergency, The People’s Hospital of Suzhou New District, Suzhou, 215000, China
*Address correspondence to: Jingjing Wang, wangjingjing5555@163.com
#These authors contributed equally to this work
Received: 30 July 2020; Accepted: 22 November 2020
Abstract: Oxidative stress-mediated cell death in cardiomyocytes contributes to the development of atrial fibrillation. However, the detailed mechanisms are still unclear. In the present study, we established atrial fibrillation models in mice. The cardiomyocytes were isolated from atrial fibrillation mice and normal mice and were cultured in vitro, respectively. The results showed that cell proliferation and viability in cardiomyocytes with atrial fibrillation were significantly lower than the cells from the normal mice. Consistently, atrial fibrillation cardiomyocytes were prone to suffer from apoptotic cell death. Also, the oxidative stress and ferroptosis-associated signatures were significantly increased in atrial fibrillation cardiomyocytes compared to normal cardiomyocytes, and ferroptosis inhibitor and NAC rescued cell viability in atrial fibrillation cardiomyocytes during in vitro cell culture. In addition, low-expressed miR-143-3p was observed in atrial fibrillation cardiomyocytes compared to normal cardiomyocytes, and overexpression of miR-143-3p increased cell proliferation and inhibited cell death in atrial fibrillation cardiomyocytes. Furthermore, glutamic-oxaloacetic transaminase 1 could be negatively regulated by miR-143-3p in normal cardiomyocytes, and miR-143-3p overexpression inhibited cell ferroptosis in atrial fibrillation cardiomyocytes by sponging glutamic-oxaloacetic transaminase 1. Collectively, overexpression of miR-143-3p increased cell viability and promoted cell proliferation in cardiomyocytes with atrial fibrillation by inhibiting glutamic-oxaloacetic transaminase 1 mediated oxidative damages and cell ferroptosis.
Keywords: Atrial fibrillation; Ferroptosis; Cardiomyocytes; MiR-143-3p; Glutamic-oxaloacetic transaminase 1; Ferrostatin-1
As the most common cardiac arrhythmia, atrial fibrillation seriously degrades the life quality of human beings, which is associated with pronounced morbidity and mortality (Donnellan et al., 2020; Qin and Heist, 2020). The pathogenesis of atrial fibrillation is very complicated, which can be attributed to various risk factors, such as heart failure (Thoren et al., 2020), hypertension (Kallistratos et al., 2018), and mitral valve disease (Bagge and Blomstrom-Lundqvist, 2018). Recent data identified that atrial fibrillation progression was closely related to oxidative stress-induced damages on cardiomyocytes (Zhang et al., 2018), and atrial fibrillation was featured by cardiomyocyte death (Freundt et al., 2018). Hence, uncovering the underlying mechanisms of oxidative damages induced cardiomyocyte death might give some insights into the molecular mechanisms of atrial fibrillation progression. Ferroptosis is an iron-dependent oxidative form of cell death and characterized by the accumulation of lipid ROS, iron and mitochondrial superoxide, and aberrant alterations in mitochondrial membrane potential (Kraft et al., 2020; Yang and Chi, 2020). Previous data suggested that oxidative stress induced various types of cell death, including necroptosis (Ferrada et al., 2020) and ferroptosis (Lewerenz et al., 2018). Of note, Wu et al. (2018) found that oxidative stress induced cell ferroptosis in PC12 cells, and Totsuka et al. (2019) evidenced that oxidative stress induced ferroptotic cell death in retinal pigment epithelial cells. Besides, cell ferroptosis involved in the development of multiple diseases, such as Parkinson’s disease (Guiney et al., 2017), pulmonary disease (Minagawa et al., 2020), and heart disease (Del Re et al., 2019).
MicroRNAs (miRNAs) are a group of small non-coding RNAs, which regulate cell functions by acting as post-transcriptional regulators to competitively bind to the 3’ untranslated regions (UTR) of target genes (Javanmard et al., 2020; Yang et al., 2020). Recent data implicated that miRNAs played important roles in the regulation of atrial fibrillation development (Min et al., 2020; Wang et al., 2019c). For example, miR-32d inhibited apoptosis in cardiomyocytes with atrial fibrillation (Liu et al., 2019), and overexpression of miR-27b-3p attenuated atrial fibrosis in rats with atrial fibrillation (Lv et al., 2019). In addition, miRNAs were closely related to oxidative stress (Wang et al., 2020a; Zhang et al., 2020) and cell ferroptosis (Niu et al., 2019; Zhang et al., 2018). Therefore, miRNAs might serve as novel prognostic and therapeutic agents for atrial fibrillation. Interestingly, recent data evidenced that miR-143-3p involved in regulating cardiomyocyte proliferation and death in mice models with myocardial infarction (Ghavami et al., 2018; Nam et al., 2019), and our preliminary experiments also supported that miR-143-3p was significantly downregulated in cardiomyocytes with atrial fibrillation compared to normal cardiomyocytes, suggesting that the expression patterns of miR-143-3p were altered in atrial fibrillation cardiomyocytes cells. Besides, miR-143-3p regulated multiple cell functions, such as cell proliferation (Xia et al., 2018), migration (Yang et al., 2018), apoptosis (Han et al., 2018), and oxidative stress (Yu et al., 2018), hence, miR-143-3p was selected for further analysis in this study.
Glutamic-oxaloacetic transaminase 1 is crucial for cellular glutaminolysis, which can convert glutamate (Glu) into a-KG and promotes ROS generation (Zhang et al., 2018). Currently, the role of glutamic-oxaloacetic transaminase 1 in the regulation of cell functions is controversial (Wang et al., 2019a; Zhang et al., 2018). On the one hand, glutamic-oxaloacetic transaminase 1 increases cell viability by sustaining normal glutamine metabolism (Wang et al., 2019a). On the other, a recent study also verified that glutamic-oxaloacetic transaminase 1 promotes melanoma cell ferroptosis by triggering oxidative damages (Zhang et al., 2018). Our preliminary results suggested that glutamic-oxaloacetic transaminase 1 was increased in atrial fibrillation cardiomyocytes and Erastin-treated normal cardiomyocytes compared to normal cardiomyocytes, indicating that glutamic-oxaloacetic transaminase 1 might contribute to atrial fibrillation progression by regulating cardiomyocyte functions. However, the detailed mechanisms are still needed to be elucidated. Furthermore, glutamic-oxaloacetic transaminase 1 could be regulated by miRNAs (Wang et al., 2019a; Zhang et al., 2018), and the online starBase software (http://starbase.sysu.edu.cn/) predicted that glutamic-oxaloacetic transaminase 1 was the downstream target of miR-143-3p.
Collectively, this study aimed to investigate the role of miR-143-3p/GOT glutamic-oxaloacetic transaminase 1 axis in the regulation of atrial fibrillation pathogenesis, which will shed light on the discovery of new diagnostic and therapeutic agents for atrial fibrillation treatment in the clinic.
Induction of atrial fibrillation mice models
Wild-type (WT) C57BL/6 mice (N = 20, weight from 20 to 25 g, 8 weeks age) were obtained and divided into two groups equally. All the mice were fed and maintained under the conditions with temperature 23 ± 2°C, humidity 55 ± 5%, 12:12 h light/dark cycle, in the Animal Research Center of Suzhou High-tech Zone People’s Hospital. The atrial fibrillation mice models were established by using the methods mentioned in the previous study (Lu et al., 2010). Briefly, the mice were anesthetized by intravenously injecting Barbiturate at the concentration of 100 mg/kg, and the standards for successful euthanasia included: (1) No cardiac arrest; (2) No spontaneous breath for at least 3 min; and (3) No blinking reflex in mice. Then, the mice were injected with 40 mg/kg sodium pentobarbital to establish atrial fibrillation mice models. All the animal experiments were approved by the Animal Care and Use Committee of the Suzhou High-tech Zone People’s Hospital. The mice suffered from rapid irregular atrial rhythm for at least one second were regarded as atrial fibrillation mice.
The cardiomyocytes were isolated from atrial fibrillation-mice and normal mice heart ventricles according to the procedures from a previous study (Liu et al., 2019). Briefly, the mice chests were opened, and the hearts were removed and excised into pieces with about 1 mm3 volume. Next, the above heart tissues were treated with trypsin and type II collagenase for 2 h at 37°C for digestion. After differential adherence, the cells were harvested, and the cardiomyocytes were collected and cultured in the Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) containing 10% fetal bovine serum (FBS, Gibco, USA). In addition, the HEK-293T cells were purchased from ATCC (Manassas, Virginia, USA) and were also cultivated in the same medium. The cells were placed in an incubator with a condition of 5% CO2 at 37°C, and the above cardiomyocytes were prepared for further analysis at 48 h post-culture when the cell confluency reached about 70–80%.
The miR-NC (5’-GAG CUA CAG UGC UUC AUC UCA-3’), miR-143-3p mimic (5’-UGA GAU GAA GCA CUG UAG CUC-3’), and inhibitor (5’-GAG CUA CAG UGC UUC AUC UCA-3’) were designed and constructed by Sangon Biotech (Shanghai, China). The glutamic-oxaloacetic transaminase 1 overexpression vectors were purchased from Vigene Biosciences (Shanghai, China). The shRNA vectors for glutamic-oxaloacetic transaminase 1 (5’-CCG GGC GTT GGT ACA ATG GAA CAA ACT CGA GTT TGT TCC ATT GTA CCA ACG CTT TTT G-3’) were obtained from GeneChem (Shanghai, China). The vectors were delivered into the cells by using a Lipofectamine 2000 transfection kit (Invitrogen, USA) according to the manufacturer’s instruction.
Total RNA was extracted from the cardiomyocytes isolated from normal and atrial fibrillation mice by using the commercial TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. Real-Time qPCR was conducted to evaluate gene expressions at transcriptional levels based on the procedures in the previous study (Ferrada et al., 2020). Specifically, total RNAs were reversely transcribed into complementary DNA (cDNA) by using the commercial AmpliTaq DNA Polymerase reagent (Life Technologies, USA), and the 2% agarose gel electrophoresis assay was performed to validate that we had successfully obtained the PCR products. Next, the One-Step TB Green TM PrimeScript TM RT-PCR kit (Takara, Japan) was used to quantify the relative expression levels of miR-143-3p and glutamic-oxaloacetic transaminase 1 mRNA. Of note, miR-143-3p was normalized by U6, and the glutamic-oxaloacetic transaminase 1 mRNA was normalized by β-actin. The primer sequences for the associated genes were listed as follows: miR-143-3p (Forward: 5’-TGA GAT GAA GCA CTG TAG CTC-3’, Reverse: 5’-GCT ACA GTG CTTCATCTC ATT-3’) (Wang et al., 2020b), U6(Forward: 5’-CTC GCT TCG GCA GCA CA-3’, Reverse: 5’-AAC GCT TCA CGA ATT TGC GT), β-actin ( Forward: 5’-CGT AAA GAC CTC TAT GCC AAC A-3’, Reverse: 5’-GGA GGA GCA ATG ATC TTG ATC T-3’) (Hu et al., 2018) and glutamic-oxaloacetic transaminase 1 (Forward: 5’- TGC TAC TGG GAT GCG GAG AAG A-3’, Reverse: 5’-TGC ATG ACA GCA GCG ATC TGC T-3’) (Sun et al., 2020).
Total proteins were extracted from the normal cardiomyocytes and atrial fibrillation cardiomyocytes by using the RIPA lysis buffer (Beyotime, Shanghai, China) in keeping with the manufacturer’s instruction. The expression levels of the proteins were detected by using Western Blot according to the procedures provided by the previous study (Ferrada et al., 2020). The primary antibodies against glutamic-oxaloacetic transaminase 1 (1:1500, #ab221939, Abcam, UK) and β-actin (1:2000, #ab6276, Abcam, UK). The secondary antibody against horseradish peroxidase-conjugated secondary antibody (1:2000, #G-21040, Invitrogen, USA).
Cell Counting Kit-8 for cell proliferation
The normal cardiomyocytes and atrial fibrillation cardiomyocytes were cultured under the standard conditions for 0, 24, 48, 72, and 96 h, respectively. The commercial CCK-8 kit (AbMole, USA) was employed to measure cell proliferation abilities according to the manufacturer’s protocol. Briefly, the cells were seeded into the 96-well plates, and the CCK-8 reaction solution was incubated with the cells at the concentration of 20 μL/well for 4 h at room temperature. After that, the optical density values were measured at the wavelength of 450 nm to evaluate cell proliferation abilities.
Trypan Blue staining assay for cell viability
The cells were administered with different treatments and vectors transfection. After that, cells were cultured under the standard culture conditions for 0, 24, 48, 72, and 96 h, respectively. The cells were harvested and stained with trypan blue staining buffer (Invitrogen, USA) in keeping with the manufacturer’s instructions. The dead blue cells were counted under the light microscope. The following formula was used to calculate cell viability (%) = (total cells – dead cells)/total cells.
Flow cytometry for cell apoptosis
The cells were harvested and double-stained with Annexin V-FITC and propidium iodide (PI) by using a cell apoptosis kit (Thermo Fisher Scientific, MA, USA) to evaluate cell apoptosis ratio. The early apoptotic cells were stained with Annexin V-FITC alone, the late apoptotic cells were double-stained with Annexin V-FITC and PI, and the necroptotic cells were stained with PI alone. The above cells were counted by using FCM purchased from BD Biosciences (CA, USA).
2,7-Dichlorodi-hydrofluorescein diacetate probes
The commercial Reactive Oxygen Species assay kit (Invitrogen, USA) containing DCFH-DA probes were employed to detect Reactive Oxygen Species levels normal cardiomyocytes and atrial fibrillation cardiomyocytes. The normal cardiomyocytes and atrial fibrillation cardiomyocytes were collected and incubated with DCFH-DA probes for 30 min at room temperature in darkness according to the procedures provided by the manufacturer. The DCFH-DA probes were catalyzed into DCF with fluorescence, and the intensity of fluorescence was photographed at the wavelength of 525 nm (maximum emission) and 480 nm (maximum excitation) by using a fluorescence microscope (Leica, Germany). In addition, the ROS levels were quantified by using Image J software.
The lipid ROS levels in normal cardiomyocytes and atrial fibrillation cardiomyocytes were detected according to the previous study (Wang et al., 2019b). In brief, the cells were incubated with C11-BODIPY (Thermo Fisher, USA) for 30 min at 37°C without light. After that, the cell samples were washed with PBS buffer, and the flow cytometer (BD Biosciences, CA, USA) was employed to measure the fluorescence at the simultaneous acquisition of green (485/510 nm) and red signals (516/610 nm) to reflect lipid ROS levels in cells.
Mitochondrial superoxide measurements
The cells were collected and incubated with the fluorescence probes specific for mitochondrial superoxide, and the MitoSOXTM Red Mitochondrial Superoxide Indicator for live-cell imaging (Invitrogen, USA) was employed to monitor mitochondrial superoxide production according to the previous study (Wang et al., 2019b). Cellular fluorescence was measured at an excitation/emission value of 510/580 nm, and Image J software was used to quantify the fluorescence intensity to reflect the levels of mitochondrial superoxide.
Mitochondrial membrane potential measurements
The Mitochondrial Membrane Potential Kit MAK-159 (Sigma, USA) was used to detect mitochondrial membrane potential according to the previous study (Wang et al., 2019b). Briefly, the cells were seeded into the 96-well plates at the density of 5 × 104 cells per well and incubated with Erastin or dimethyl sulfoxide. After 48 h incubation, the cells were incubated with the reaction buffer A and B, respectively. The fluorescence intensity levels were calculated to reflect mitochondrial potential based on the protocol from the previous study (Wang et al., 2019b).
The normal cardiomyocytes and atrial fibrillation cardiomyocytes were transfected with different vectors. After that, the total iron and ferrous iron were measured by using the commercial Iron Assay kit obtained from Sigma (USA) based on the protocol provided by the manufacturer. The detailed procedures for total iron and ferrous iron measurements were explained in the previous study (Wang et al., 2019b).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay
A commercial terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay kit (Beyotime, Shanghai, China) was employed to detect cell apoptosis ratio according to the manufacturer’s instruction. Briefly, the normal cardiomyocytes and atrial fibrillation cardiomyocytes were harvested and fixed with 4% paraformaldehyde for 40 min. The PBST (containing 0.3% Triton X-100) was used to increase permeability in cell membranes. After that, the cells were stained with TUNEL reaction solution and DAPI, respectively. The above cells were observed and photographed under a fluorescence microscope. The apoptotic cells were TUNEL-positive, and all cell’ nuclei were stained blue with DAPI.
Dual-luciferase reporter gene system
The binding sites of miR-143-3p and glutamic-oxaloacetic transaminase 1 mRNA were predicted by using the online starBase software (http://starbase.sysu.edu.cn/), and the binding sites in glutamic-oxaloacetic transaminase 1 were mutated. The wild-type (Wt) and mutant (Mut) 3’ UTR regions of glutamic-oxaloacetic transaminase 1 were cloned into pMIR-REPORT vectors to generate Wt- glutamic-oxaloacetic transaminase 1 and Mut- glutamic-oxaloacetic transaminase 1 vectors, respectively. The miR-143-3p mimic, miR-NC, Wt- glutamic-oxaloacetic transaminase 1, and Mut- glutamic-oxaloacetic transaminase 1 vectors were co-transfected into HEK-293T cells, and the relative luciferase activity was determined by using a dual-luciferase reporter assay kit (Promega, USA) according to the manufacturer’s protocol.
The data were collected and analyzed by using the SPSS data analysis software (Version 18.0, IBM, USA). The data were represented as mean ± standard deviation (SD). Differences between two groups were compared by using the Student’s t-test, and the differences among multiple groups (above 2 groups) were analyzed by one-way Analysis of Variance followed by the post hoc Tukey’s test for multiple comparison correction. Each experiment was repeated at least 3 times, and p < 0.05 was regarded as statistical significance.
Cell functions were determined in cardiomyocytes isolated from atrial fibrillation mice and normal mice
Previous data suggested that the biological functions of cardiomyocytes with atrial fibrillation were altered in atrial fibrillation mice models (Lu et al., 2010); hence, we compared the differences of cardiomyocytes between atrial fibrillation cardiomyocytes and normal cardiomyocytes in terms of cell proliferation (Fig. 1A), viability (Fig. 1B) and apoptosis (Figs. 1C and 1D). To achieve this, the mice models for atrial fibrillation were inducted, and the cardiomyocytes were isolated and cultured in vitro under the standard conditions for 24, 48, 72, and 96 h, respectively. The CCK-8 assay results showed that the cell proliferation abilities of atrial fibrillation cardiomyocytes were lower than the normal cardiomyocytes (Fig. 1A). In addition, the trypan blue staining results showed that cell viability in atrial fibrillation cardiomyocytes was significantly lower than in normal cardiomyocytes (Fig. 1B). The above results were also validated by the FCM results for cell apoptosis (Figs. 1C and 1D); specifically, the cell apoptosis ratio in atrial fibrillation cardiomyocytes was much higher than that in normal cardiomyocytes (Figs. 1C and 1D). Besides, most of the dead atrial fibrillation cardiomyocytes suffered from late apoptosis, instead of early apoptosis and necroptosis, during in vitro cell culture (Figs. 1C and 1D). The above results suggested that atrial fibrillation caused damages to cardiomyocytes and influenced their normal biological functions.
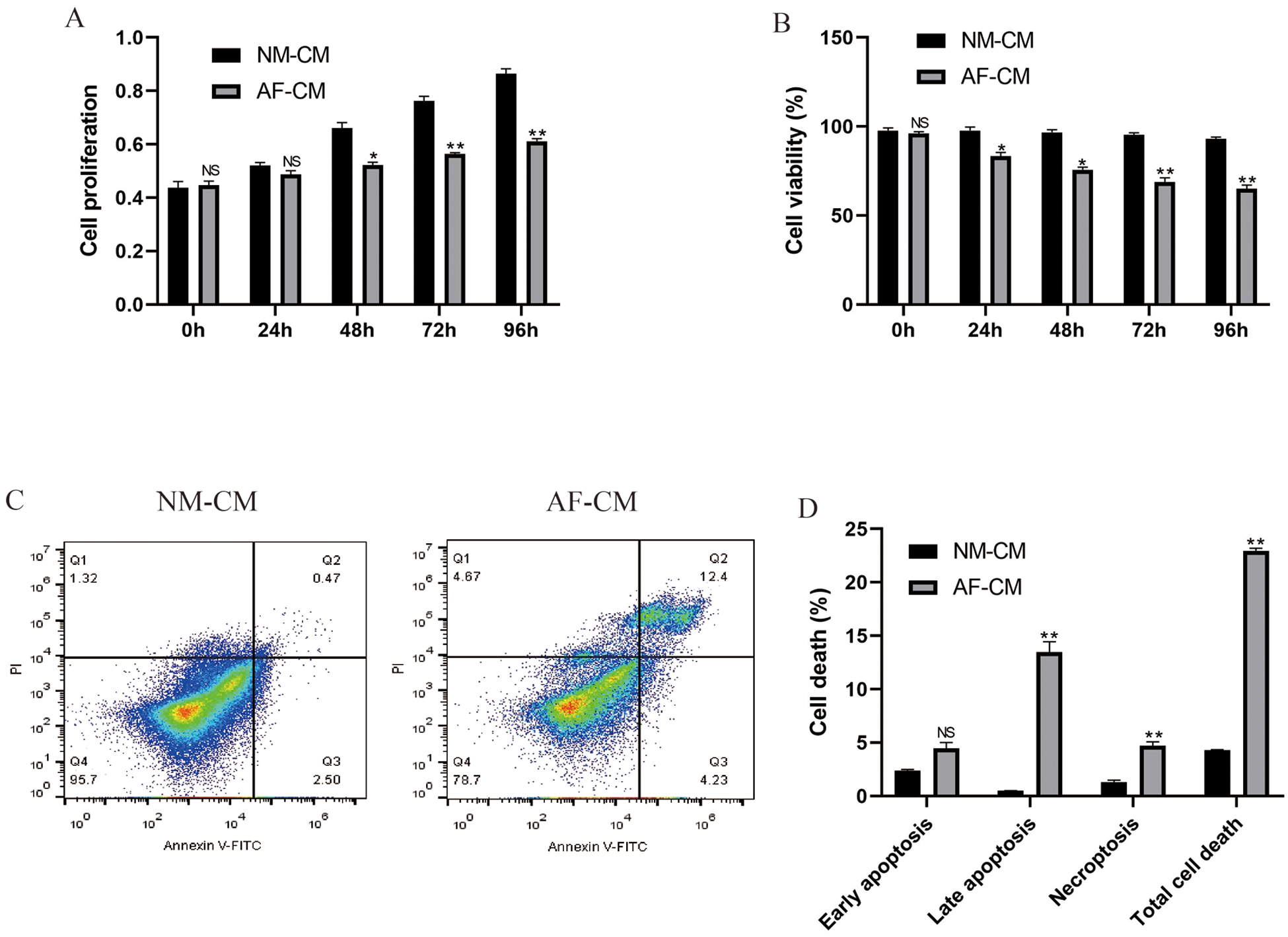
Figure 1: Differential cell functions of normal cardiomyocytes and atrial fibrillation cardiomyocytes during in vitro culture.
Detection of oxidative stress mediated ferroptosis indicators in atrial fibrillation cardiomyocytes and normal cardiomyocytes
Oxidative stress was closely related to atrial fibrillation progression; hence, we next investigated whether atrial fibrillation influenced oxidative stress in cardiomyocytes. The DCFH-DA staining results showed that ROS levels were significantly increased in atrial fibrillation cardiomyocytes compared to normal cardiomyocytes (Figs. 2A and 2B). In addition, the lipid ROS levels were also elevated in atrial fibrillation cardiomyocytes compared to normal cardiomyocytes after 48 h in vitro culture (Fig. 2C). Since oxidative stress-mediated cell ferroptosis was characterized by upregulation of lipid ROS, we explored whether the indicators for cell ferroptosis could be observed in atrial fibrillation cardiomyocytes. The results showed that intracellular concentrations of total iron (Fig. 2F), Fe2+ (Fig. 2G), and mitochondrial superoxide (Fig. 2D) were all elevated in atrial fibrillation cardiomyocytes compared to normal cardiomyocytes. Consistently, the decreased mitochondrial membrane potential was observed in atrial fibrillation cardiomyocytes instead of normal cardiomyocytes (Fig. 2E), indicating that oxidative damages and cell ferroptosis appeared in atrial fibrillation cardiomyocytes.
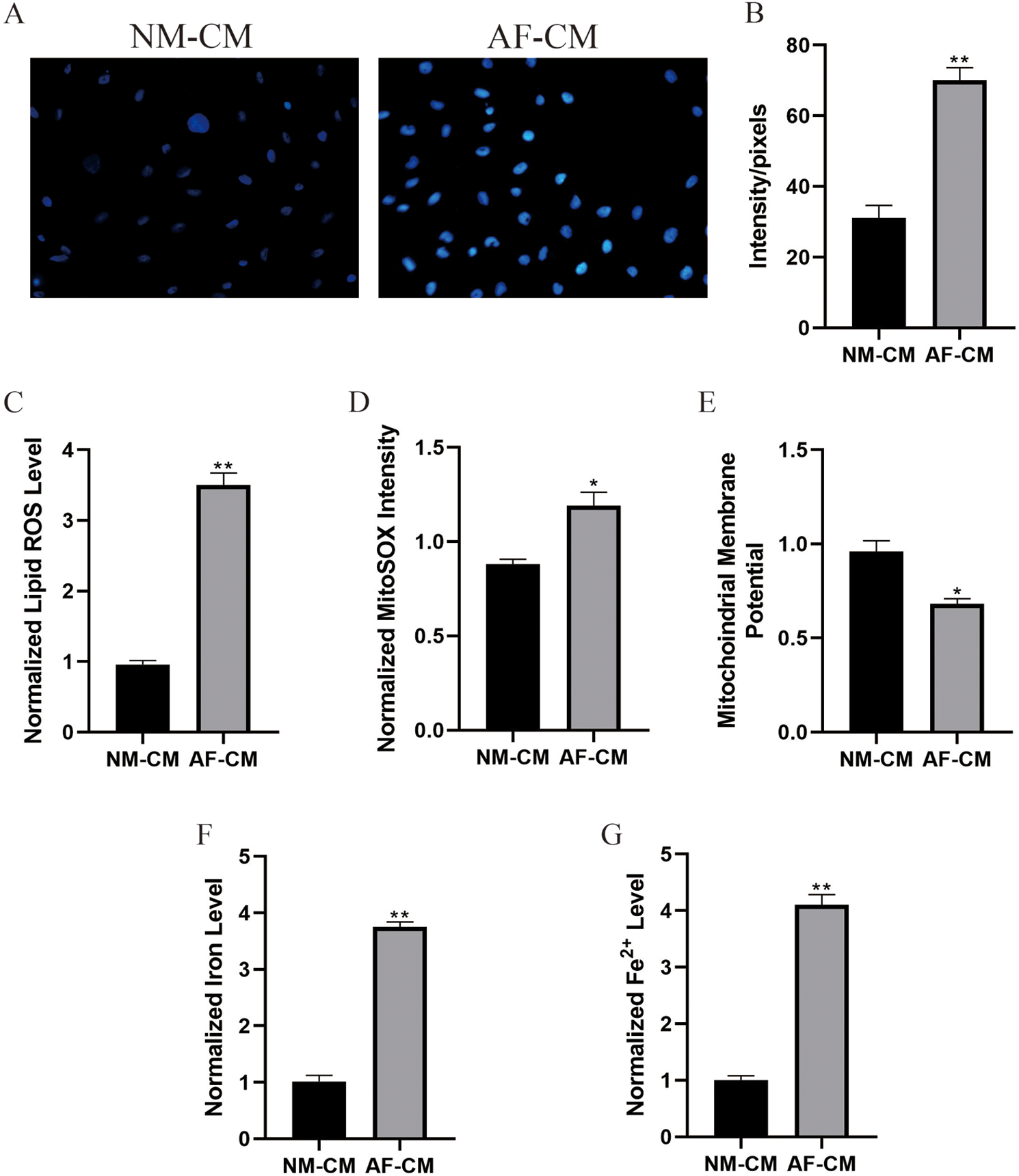
Figure 2: Observation of ferroptosis indicators in normal cardiomyocytes and atrial fibrillation cardiomyocytes at 48 h post in vitro culture.
The effects of ferrostatin-1 and NAC on cell viability in atrial fibrillation cardiomyocytes
To investigate the role of oxidative stress and ferroptosis in the regulation of the biological functions of atrial fibrillation cardiomyocytes, the cells were treated with ferroptosis inhibitor (ferrostatin-1, Fer-1) and ROS scavenger (NAC), respectively. The CCK-8 results showed that atrial fibrillation cardiomyocytes proliferation was largely limited during in vitro culture, which was promoted by both ferrostatin-1 and NAC treatments (Fig. 3A). Besides, ferrostatin-1 and NAC also rescued cell viability in atrial fibrillation cardiomyocytes (Fig. 3B). Furthermore, the apoptosis ratio of atrial fibrillation cardiomyocytes was significantly decreased by treating cells with ferrostatin-1 and NAC (Figs. 3C and 3D). In addition, we investigated the regulating mechanisms of oxidative stress and ferroptosis, and the results showed that NAC decreased lipid ROS (Fig. 3E), mitochondrial superoxide (Fig. 3F), intracellular concentrations of total iron (Fig. 3H) and Fe2+ (Fig. 3I), while increased mitochondrial membrane potential (Fig. 3G) in atrial fibrillation cardiomyocytes, suggesting that oxidative stress-induced ferroptosis was observed in atrial fibrillation cardiomyocytes.
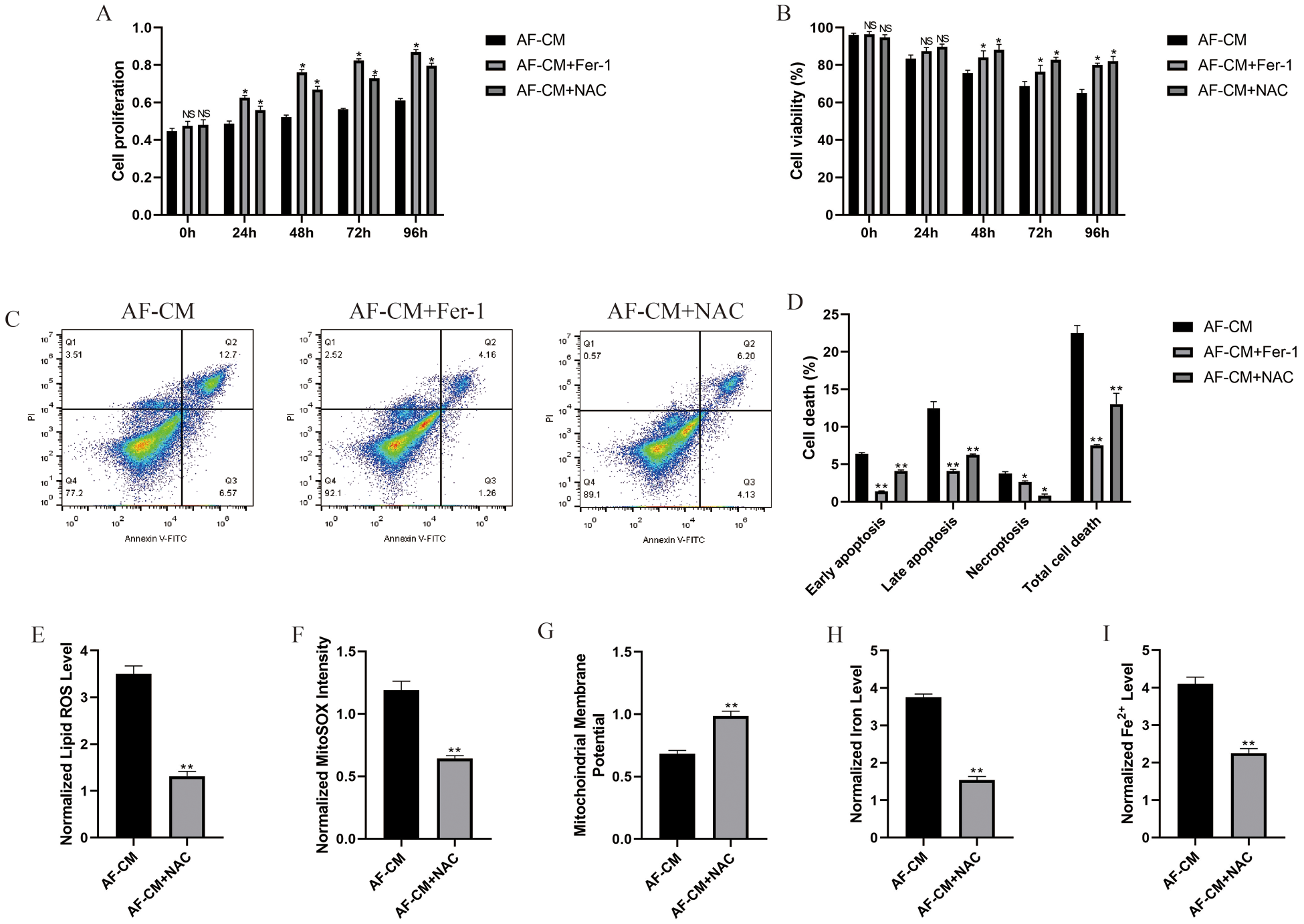
Figure 3: Ferrostatin-1 and NAC influenced cell functions in atrial fibrillation cardiomyocytes.
MiR-143-3p involved in the regulation of cell viability in atrial fibrillation cardiomyocytes
Further experiments were conducted to explore the underlying mechanisms. The Real-Time qPCR results showed that miR-143-3p was low-expressed in atrial fibrillation cardiomyocytes compared to normal cardiomyocytes (Fig. 4A); hence, miR-143-3p was overexpressed in atrial fibrillation cardiomyocytes (Fig. 4B) and downregulated in normal cardiomyocytes (Fig. 4C) to validate its biological functions in the further experiments. The CCK-8 assay results showed that miR-143-3p overexpression rescued cell proliferation abilities of atrial fibrillation cardiomyocytes in vitro (Fig. 4D), and knock-down of miR-143-3p significantly inhibited normal cardiomyocytes proliferation (Fig. 4E). Further trypan blue staining results validated that overexpression of miR-143-3p increased atrial fibrillation cardiomyocytes viability (Fig. 4F), and knock-down of miR-143-3p promoted normal cardiomyocyte death (Fig. 4G). Consistently, the TUNEL assay results showed that overexpressed miR-143-3p decreased cell apoptosis ratio in atrial fibrillation cardiomyocytes (Figs. 4H and 4I), and the FCM results showed that silencing of miR-143-3p promoted cell apoptosis in normal cardiomyocytes (Figs. 4J and 4K).
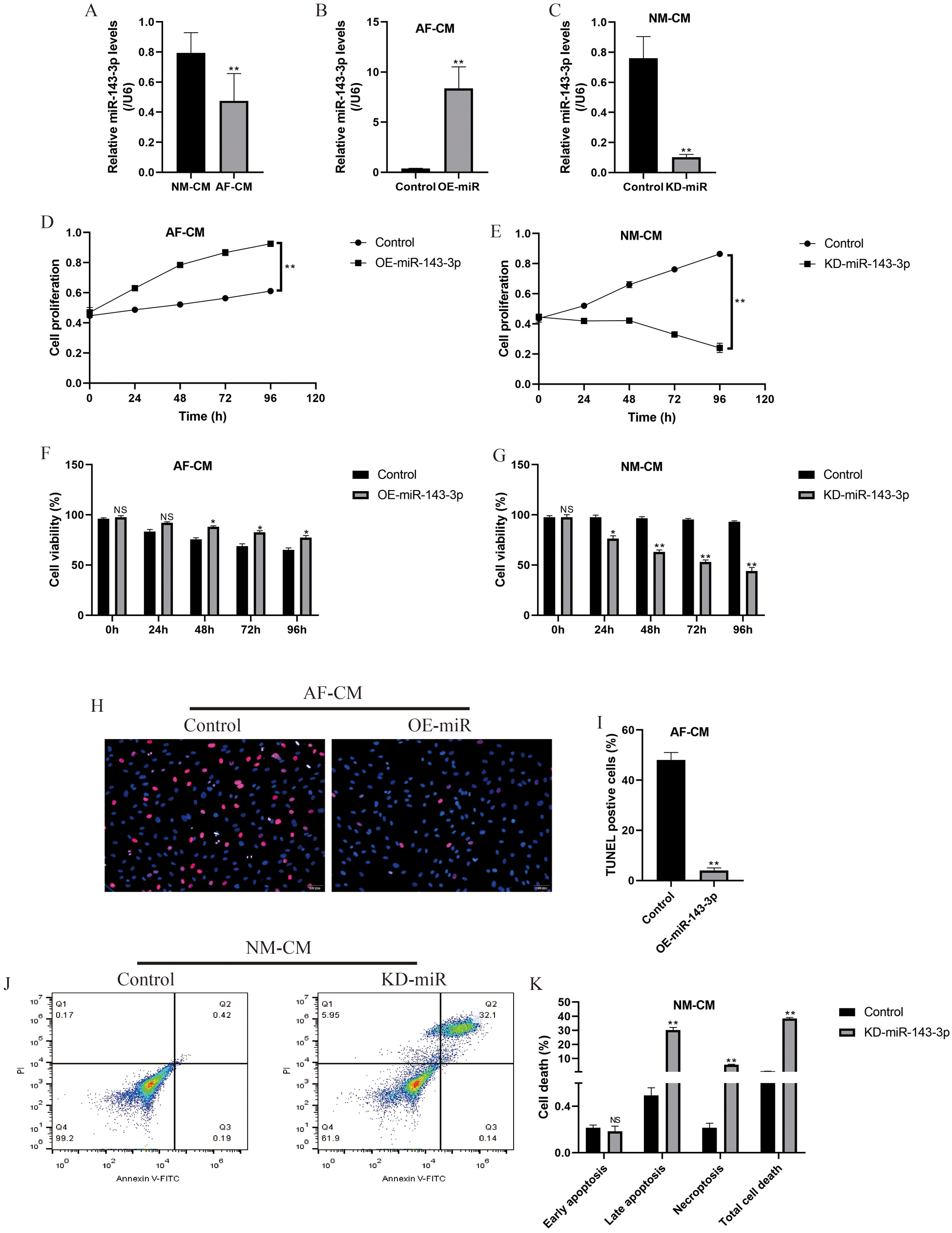
Figure 4: Influences of miR-143-3p on cell functions in normal cardiomyocytes and atrial fibrillation cardiomyocytes.
MiR-143-3p regulated cell ferroptosis in atrial fibrillation cardiomyocytes and Erastin-treated normal cardiomyocytes through targeting glutamic-oxaloacetic transaminase 1
Glutamic-oxaloacetic transaminase-1 promoted cell ferroptosis (Zhang et al., 2018), which was also validated in this study. Specifically, the expression levels of glutamic-oxaloacetic transaminase 1 were higher in atrial fibrillation cardiomyocytes (Figs. 5A and 5C; Fig. S1A) and Erastin-treated normal cardiomyocytes cells (Figs. 5B and 5C; Fig. S1A) compared to the normal cardiomyocytes. Besides, downregulation of glutamic-oxaloacetic transaminase 1 promoted atrial fibrillation cardiomyocytes proliferation (Figs. 5D and 5E) and alleviated Erastin-induced cell death in normal cardiomyocytes (Figs. 5F and 5G). Interestingly, the online starBase software (http://starbase.sysu.edu.cn/) predicted that glutamic-oxaloacetic transaminase 1 was the downstream target of miR-143-3p (Fig. 5H), which were validated by the Dual-luciferase reporter gene system results (Fig. 5I). Further results validated that glutamic-oxaloacetic transaminase 1 could be negatively regulated by miR-143-3p in normal cardiomyocytes (Fig. 5J; Fig. S1B). In addition, overexpression of miR-143-3p inhibited lipid ROS production (Fig. 5K), mitochondrial superoxide (Fig. 5L), intracellular concentrations of total iron (Fig. 5N) and Fe2+ (Fig. 5O) and increased mitochondrial membrane potential (Fig. 5M) in atrial fibrillation cardiomyocytes, which were all abrogated by upregulating glutamic-oxaloacetic transaminase 1 (Figs. 5K–5O).
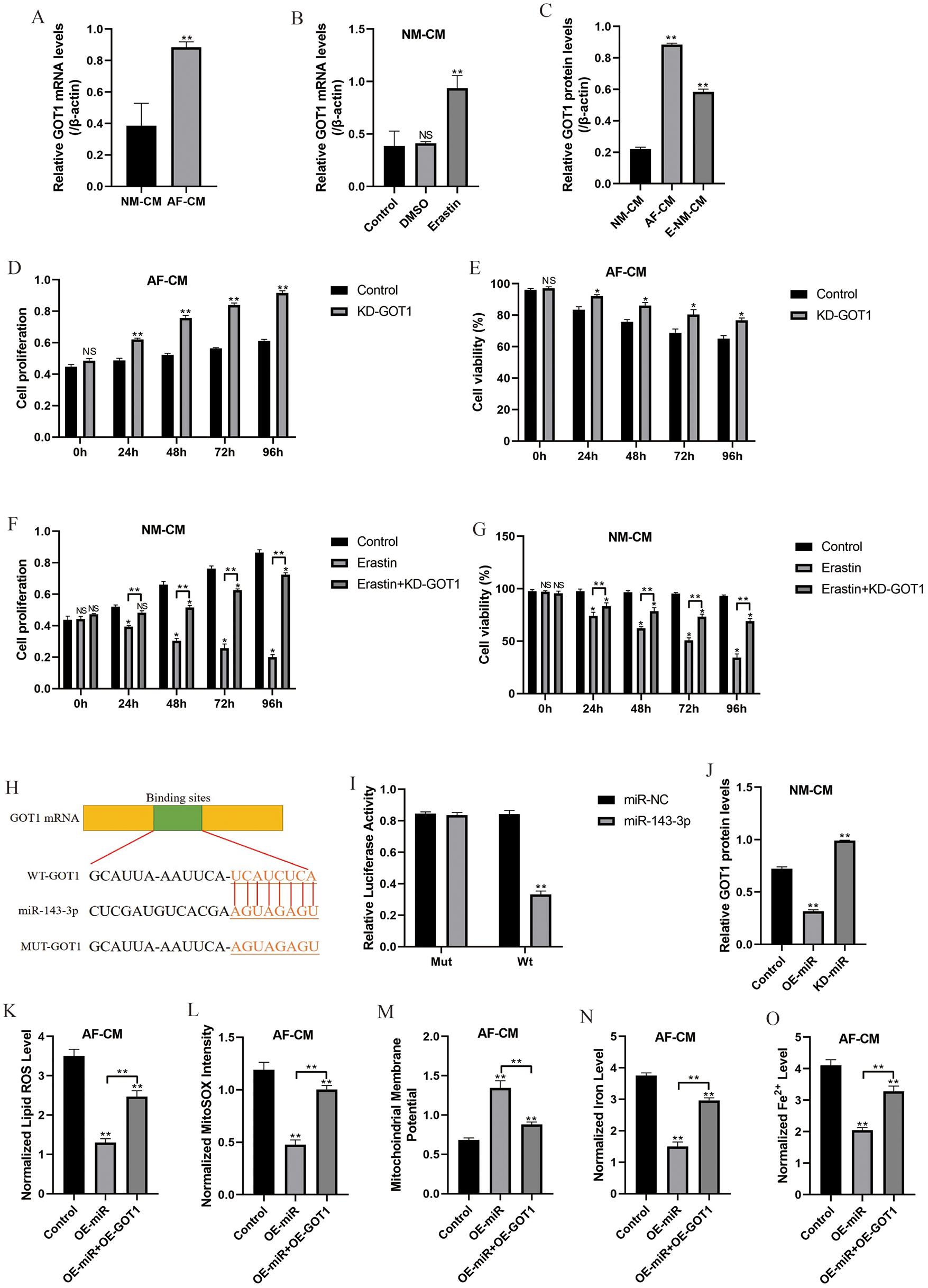
Figure 5: MiR-143-3p regulated cell proliferation, viability, and ferroptosis by targeting glutamic-oxaloacetic transaminase 1.
A lack of prognostic and therapeutic agents for atrial fibrillation seriously limits its treatment in the clinic (Donnellan et al., 2020; Qin and Heist, 2020), and uncovering the underlying mechanisms of atrial fibrillation pathogenesis might help to solve this problem. Previous data indicated that cardiomyocytes could be used as in vitro models for atrial fibrillation research (Liu et al., 2019), and cardiomyocyte death contributed to atrial fibrillation progression (Freundt et al., 2018). Hence this study established atrial fibrillation mice models, and the cardiomyocytes were isolated from mice’s heart ventricles according to the previous study (Liu et al., 2019). After in vitro culture for differential time points, we found that cell proliferation was largely limited, and cell viability was significantly decreased in the cardiomyocytes with atrial fibrillation in a time-dependent manner, which suggested that atrial fibrillation caused cell death in cardiomyocytes. Recent data also suggested that oxidative stress was closely related to atrial fibrillation progression (Zhang et al., 2018), and this study validated that ROS levels were increased in atrial fibrillation cardiomyocytes. Besides, since ferroptosis was characterized by lipid ROS accumulation (Kraft et al., 2020; Lewerenz et al., 2018; Yang and Chi, 2020), we proved that cell ferroptosis appeared in atrial fibrillation cardiomyocytes. In addition, both ferroptosis inhibitor (ferrostatin-1) and ROS scavenger (NAC) rescued atrial fibrillation cardiomyocytes viability in vitro, and NAC inhibited cell ferroptosis in atrial fibrillation cardiomyocytes, suggesting that oxidative damages mediated ferroptosis contributed to cell death in cardiomyocytes with atrial fibrillation, which were supported by the previous studies (Totsuka et al., 2019; Wu et al., 2018).
MicroRNAs (miRNAs) have been reported to regulate oxidative stress, cell ferroptosis, and atrial fibrillation progression, which might be novel diagnostic and therapeutic agents for atrial fibrillation. This study found that miR-143-3p was low-expressed in atrial fibrillation cardiomyocytes compared to normal cardiomyocytes, suggesting that the expression patterns of miR-143-3p were changed by atrial fibrillation in cardiomyocytes. Previous data indicated that miR-143-3p regulated cell proliferation (Xia et al., 2018), death (Han et al., 2018), and responses to oxidative damages (Yu et al., 2018), which enlightened us to investigate the role of miR-143-3p in the regulation of cell viability in atrial fibrillation cardiomyocytes and normal cardiomyocytes. The results showed that overexpressed rescued cell viability in atrial fibrillation cardiomyocytes, while knock-down of miR-143-3p induced normal cardiomyocyte death in vitro, suggesting that miR-143-3p protected atrial fibrillation cardiomyocytes from death. In addition, miR-143-3p alleviated cell ferroptosis in atrial fibrillation cardiomyocytes. The above results suggested that overexpression of miR-143-3p alleviated oxidative damages-induced cell ferroptosis in cardiomyocytes suffered from atrial fibrillation. Although we had evidenced that miR-143-3p played a dominant role in regulating atrial fibrillation progression in vitro, more experiments are still needed to investigate the potential interplays between miR-143-3p with other miRNAs that synergistically contributed to atrial fibrillation pathogenesis in our future work.
Glutamic-oxaloacetic transaminase 1 plays a controversial role in the regulation of cell functions by modulating glutaminolysis in a cell type-dependent manner (Wang et al., 2019a; Zhang et al., 2018); however, the role of glutamic-oxaloacetic transaminase 1 in the regulation of cell death in cardiomyocytes with atrial fibrillation is still largely unknown. The present study found that glutamic-oxaloacetic transaminase 1 was high-expressed in atrial fibrillation cardiomyocytes and Erastin-treated normal cardiomyocytes cells compared to the normal cardiomyocytes and downregulated glutamic-oxaloacetic transaminase 1 protected atrial fibrillation cardiomyocytes and Erastin-treated normal cardiomyocytes from death, suggesting that glutamic-oxaloacetic transaminase 1 inhibited cell viability in cardiomyocytes and in accordance with the previous study (Zhang et al., 2018). In addition, we found that glutamic-oxaloacetic transaminase 1 was the downstream target and could be negatively regulated by miR-143-3p in mice cardiomyocytes. Of note, glutamic-oxaloacetic transaminase 1 promoted melanoma cell ferroptosis by triggering ROS production (Zhang et al., 2018), and this study validated that the inhibiting effects of miR-143-3p overexpression on cell ferroptosis in atrial fibrillation cardiomyocytes cells were abrogated by upregulating glutamic-oxaloacetic transaminase 1, suggesting that miR-143-3p regulated atrial fibrillation cardiomyocytes cell ferroptosis by targeting glutamic-oxaloacetic transaminase 1.
Collectively, overexpression of miR-143-3p inhibited cell ferroptosis in cardiomyocytes with atrial fibrillation by inhibiting glutamic-oxaloacetic transaminase 1, consequently alleviating the development of atrial fibrillation (Fig. 6). This in vitro study provided potential therapeutic agents for atrial fibrillation treatment in the clinic; however, more clinical data are still needed to provide more evidence to support these results.
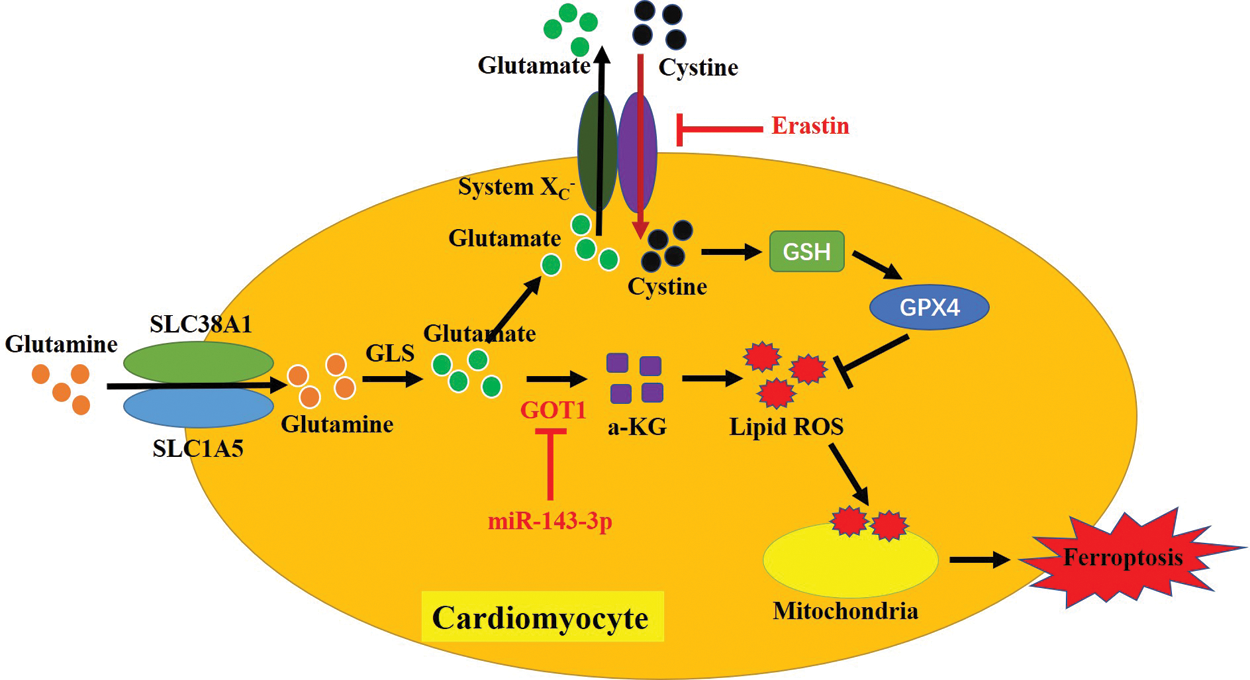
Figure 6: The graphical abstract of this study.
Availability of Data and Materials:All data generated or analyzed during this study are included in this published article.
Authors’ Contributions: Dr. Yuan Song designed and conducted all the experiments in this study and wrote the draft. Dr. Cai Wei was responsible for data collection and analysis, figures design, and language modification. Dr. Jingjing Wang financially supported this study and provided conception and guidance for this work.
Ethics Approval: All the animal experiments were approved by the Animal Care and Use Committee of the Sunzhou High-tech Zone People’s Hospital at 2020/06/18, and the approval number was No. 2020007685432.
Funding Statement: This study was financially supported by the Youth Science and Innovation Research Fund of Xinjiang Province (No. WJWY-201966).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Bagge L, Blomstrom-Lundqvist C. (2018). Surgical ablation for atrial fibrillation in mitral valve disease: Impact of the maze procedure-authors’ response. Europace 20: f458–f459. DOI 10.1093/europace/eux329. [Google Scholar] [CrossRef]
Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. (2019). Fundamental mechanisms of regulated cell death and implications for heart disease. Physiological Reviews 99: 1765–1817. DOI 10.1152/physrev.00022.2018. [Google Scholar] [CrossRef]
Donnellan E, Wazni O, Kanj M, Elshazly MB, Hussein A, Baranowski B, Hanna M, Patel D, Trulock K, Martyn M, Menon V, Saliba W, Jaber WA. (2020). Atrial fibrillation ablation in patients with transthyretin cardiac amyloidosis. Europace 22: 259–264. DOI 10.1093/europace/euz314. [Google Scholar] [CrossRef]
Ferrada L, Barahona MJ, Salazar K, Vandenabeele P, Nualart F. (2020). Vitamin C controls neuronal necroptosis under oxidative stress. Redox Biology 29: 101408. DOI 10.1016/j.redox.2019.101408. [Google Scholar] [CrossRef]
Freundt JK, Frommeyer G, Wotzel F, Huge A, Hoffmeier A, Martens S, Eckardt L, Lange PS. (2018). The transcription factor ATF4 promotes expression of cell stress genes and cardiomyocyte death in a cellular model of atrial fibrillation. BioMed Research International 2018: 3694362. DOI 10.1155/2018/3694362. [Google Scholar] [CrossRef]
Ghavami S, Yeganeh B, Zeki AA, Shojaei S, Kenyon NJ, Ott S, Samali A, Patterson J, Alizadeh J, Moghadam AR, Dixon IMC, Unruh H, Knight DA, Post M, Klonisch T, Halayko AJ. (2018). Autophagy and the unfolded protein response promote profibrotic effects of TGF-β1 in human lung fibroblasts. American Journal of Physiology-Lung Cellular and Molecular Physiology 314: L493–L504. DOI 10.1152/ajplung.00372.2017. [Google Scholar] [CrossRef]
Guiney SJ, Adlard PA, Bush AI, Finkelstein DI, Ayton S. (2017). Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochemistry International 104: 34–48. DOI 10.1016/j.neuint.2017.01.004. [Google Scholar] [CrossRef]
Han L, Tang M, Xu X, Jiang B, Wei Y, Qian H, Lu X. (2018). MiR-143-3p suppresses cell proliferation, migration, and invasion by targeting Melanoma-Associated Antigen A9 in laryngeal squamous cell carcinoma. Journal of Cellular Biochemistry 120: 1245–1257. DOI 10.1002/jcb.27084. [Google Scholar] [CrossRef]
Hu YB, Ye XT, Zhou QQ, Fu RQ. (2018). Sestrin 2 attenuates rat hepatic stellate cell (HSC) activation and liver fibrosis via an mTOR/AMPK-dependent mechanism. Cellular Physiology and Biochemistry 51: 2111–2122. DOI 10.1159/000495829. [Google Scholar] [CrossRef]
Javanmard AR, Dokanehiifard S, Bohlooli M, Soltani BM. (2020). LOC646329 long non-coding RNA sponges miR-29b-1 and regulates TGFβ signaling in colorectal cancer. Journal of Cancer Research and Clinical Oncology 146: 1205–1215. DOI 10.1007/s00432-020-03145-6. [Google Scholar] [CrossRef]
Kallistratos MS, Poulimenos LE, Manolis AJ. (2018). Atrial fibrillation and arterial hypertension. Pharmacological Research 128: 322–326. DOI 10.1016/j.phrs.2017.10.007. [Google Scholar] [CrossRef]
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kossl J, Brandner S, Daniels JD, Schmitt-Kopplin P, Hauck SM, Stockwell BR, Hadian K, Schick JA. (2020). GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Central Science 6: 41–53. DOI 10.1021/acscentsci.9b01063. [Google Scholar] [CrossRef]
Lewerenz J, Ates G, Methner A, Conrad M, Maher P. (2018). Oxytosis/Ferroptosis—(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Frontiers in Neuroscience 12: 214. DOI 10.3389/fnins.2018.00214. [Google Scholar] [CrossRef]
Liu L, Zhang H, Mao H, Li X, Hu Y. (2019). Exosomal miR-320d derived from adipose tissue-derived MSCs inhibits apoptosis in cardiomyocytes with atrial fibrillation (AF). Artificial Cells, Nanomedicine, and Biotechnology 47: 3976–3984. DOI 10.1080/21691401.2019.1671432. [Google Scholar] [CrossRef]
Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Zhang Y, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B. (2010). MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 122: 2378–2387. DOI 10.1161/CIRCULATIONAHA.110.958967. [Google Scholar] [CrossRef]
Lv X, Li J, Hu Y, Wang S, Yang C, Li C, Zhong G. (2019). Overexpression of miR-27b-3p targeting Wnt3a regulates the signaling pathway of Wnt/β-catenin and attenuates atrial fibrosis in rats with atrial fibrillation. Oxidative Medicine and Cellular Longevity 2019: 5703764. [Google Scholar]
Min J, Shen H, Wang P, Zhang Y, Yu Y, Wang Q, Wang S, Xi W, Nguyen QM, Xiao J, Wang Z. (2020). Identification and characterization of circular RNAs in atrial appendage of patients with atrial fibrillation. Experimental Cell Research 389: 111821. DOI 10.1016/j.yexcr.2020.111821. [Google Scholar] [CrossRef]
Minagawa S, Yoshida M, Araya J, Hara H, Imai H, Kuwano K. (2020). Regulated necrosis in pulmonary disease: A focus on necroptosis and ferroptosis. American Journal of Respiratory Cell and Molecular Biology 62: 554–562. DOI 10.1165/rcmb.2019-0337TR. [Google Scholar] [CrossRef]
Nam SA, Kim WY, Kim JW, Kang MG, Park SH, Lee MS, Kim HW, Yang CW, Kim J, Kim YK. (2019). Autophagy in FOXD1 stroma-derived cells regulates renal fibrosis through TGF-β and NLRP3 inflammasome pathway. Biochemical and Biophysical Research Communications 508: 965–972. DOI 10.1016/j.bbrc.2018.11.090. [Google Scholar] [CrossRef]
Niu Y, Zhang J, Tong Y, Li J, Liu B. (2019). Physcion 8-O-β-glucopyranoside induced ferroptosis via regulating miR-103a-3p/GLS2 axis in gastric cancer. Life Sciences 237: 116893. DOI 10.1016/j.lfs.2019.116893. [Google Scholar] [CrossRef]
Qin D, Heist EK. (2020). Atrial fibrillation ablation in congestive heart failure with preserved ejection fraction: Tackling the vicious twins. Journal of Cardiovascular Electrophysiology 31: 689–691. DOI 10.1111/jce.14370. [Google Scholar] [CrossRef]
Sun L, Fu J, Lin SH, Sun JL, Xia L, Lin CH, Liu L, Zhang C, Yang L, Xue P, Wang X, Huang S, Han X, Chen HL, Huang MS, Zhang X, Huang SK, Zhou Y. (2020). Particulate matter of 2.5 μm or less in diameter disturbs the balance of TH17/regulatory T cells by targeting glutamate oxaloacetate transaminase 1 and hypoxia-inducible factor 1α in an asthma model. Journal of Allergy and Clinical Immunology 145: 402–414. DOI 10.1016/j.jaci.2019.10.008. [Google Scholar] [CrossRef]
Thoren E, Wernroth ML, Christersson C, Grinnemo KH, Jideus L, Stahle E. (2020). Compared with matched controls, patients with postoperative atrial fibrillation (POAF) have increased long-term AF after CABG, and POAF is further associated with increased ischemic stroke, heart failure and mortality even after adjustment for AF. Clinical Research in Cardiology 109: 1232–1242. DOI 10.1007/s00392-020-01614-z. [Google Scholar] [CrossRef]
Totsuka K, Ueta T, Uchida T, Roggia MF, Nakagawa S, Vavvas DG, Honjo M, Aihara M. (2019). Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Experimental Eye Research 181: 316–324. DOI 10.1016/j.exer.2018.08.019. [Google Scholar] [CrossRef]
Wang J, Wang B, Ren H, Chen W (2019a). miR-9-5p inhibits pancreatic cancer cell proliferation, invasion and glutamine metabolism by targeting GOT1. Biochemical and Biophysical Research Communications 509: 241–248. DOI 10.1016/j.bbrc.2018.12.114. [Google Scholar] [CrossRef]
Wang L, Chen X, Wang Y, Zhao L, Zhao X, Wang Y (2020a). MiR-30c-5p mediates the effects of panax notoginseng saponins in myocardial ischemia reperfusion injury by inhibiting oxidative stress-induced cell damage. Biomedicine & Pharmacotherapy 125: 109963. DOI 10.1016/j.biopha.2020.109963. [Google Scholar] [CrossRef]
Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, Shi Y, Chen L, Xiao D, Yu F, Wang X, Zhou H, Cao Y, Liu S, Yan Q, Tao Y, Zhang B (2019b). Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death & Differentiation 26: 2329–2343. DOI 10.1038/s41418-019-0304-y. [Google Scholar] [CrossRef]
Wang S, Min J, Yu Y, Yin L, Wang Q, Shen H, Yang J, Zhang P, Xiao J, Wang Z (2019c). Differentially expressed miRNAs in circulating exosomes between atrial fibrillation and sinus rhythm. Journal of Thoracic Disease 11: 4337–4348. DOI 10.21037/jtd. [Google Scholar] [CrossRef]
Wang Y, Li H, Shi Y, Wang S, Xu Y, Li H, Liu D (2020b). miR-143-3p impacts on pulmonary inflammatory factors and cell apoptosis in mice with mycoplasmal pneumonia by regulating TLR4/MyD88/NF-κB pathway. Bioscience Reports 40: BSR20193419. DOI 10.1042/BSR20193419. [Google Scholar] [CrossRef]
Wu C, Zhao W, Yu J, Li S, Lin L, Chen X. (2018). Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Scientific Reports 8: 574. DOI 10.1038/s41598-017-18935-1. [Google Scholar] [CrossRef]
Xia C, Yang Y, Kong F, Kong Q, Shan C. (2018). MiR-143-3p inhibits the proliferation, cell migration and invasion of human breast cancer cells by modulating the expression of MAPK7. Biochimie 147: 98–104. DOI 10.1016/j.biochi.2018.01.003. [Google Scholar] [CrossRef]
Yang WH, Chi JT. (2020). Hippo pathway effectors YAP/TAZ as novel determinants of ferroptosis. Molecular & Cellular Oncology 7: 1699375. DOI 10.1080/23723556.2019.1699375. [Google Scholar] [CrossRef]
Yang Y, Song S, Meng Q, Wang L, Li X, Xie S, Chen Y, Jiang X, Wang C, Lu Y, Xin X, Pu H, Gui X, Li T, Xu J, Li J, Jia S, Lu D. (2020). miR24-2 accelerates progression of liver cancer cells by activating Pim1 through tri-methylation of Histone H3 on the ninth lysine. Journal of Cellular and Molecular Medicine 24: 2772–2790. DOI 10.1111/jcmm.15030. [Google Scholar] [CrossRef]
Yang Z, Wang J, Pan Z, Zhang Y. (2018). miR-143-3p regulates cell proliferation and apoptosis by targeting IGF1R and IGFBP5 and regulating the Ras/p38 MAPK signaling pathway in rheumatoid arthritis. Experimental and Therapeutic Medicine 15: 3781–3790. [Google Scholar]
Yu B, Zhao Y, Zhang H, Xie D, Nie W, Shi K. (2018). Inhibition of microRNA-143-3p attenuates myocardial hypertrophy by inhibiting inflammatory response. Cell Biology International 42: 1584–1593. DOI 10.1002/cbin.11053. [Google Scholar] [CrossRef]
Zhang K, Wu L, Zhang P, Luo M, Du J, Gao T, O’connell D, Wang G, Wang H, Yang Y. (2018). miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Molecular Carcinogenesis 57: 1566–1576. [Google Scholar]
Zhang Y, Wang C, Lu J, Jin Y, Xu C, Meng Q, Liu Q, Dong D, Ma X, Liu K, Sun H. (2020). Targeting of miR-96-5p by catalpol ameliorates oxidative stress and hepatic steatosis in LDLr-/- mice via p66shc/cytochrome C cascade. Aging 12: 2049–2069. [Google Scholar]
Supplementary Figures and Figure legends
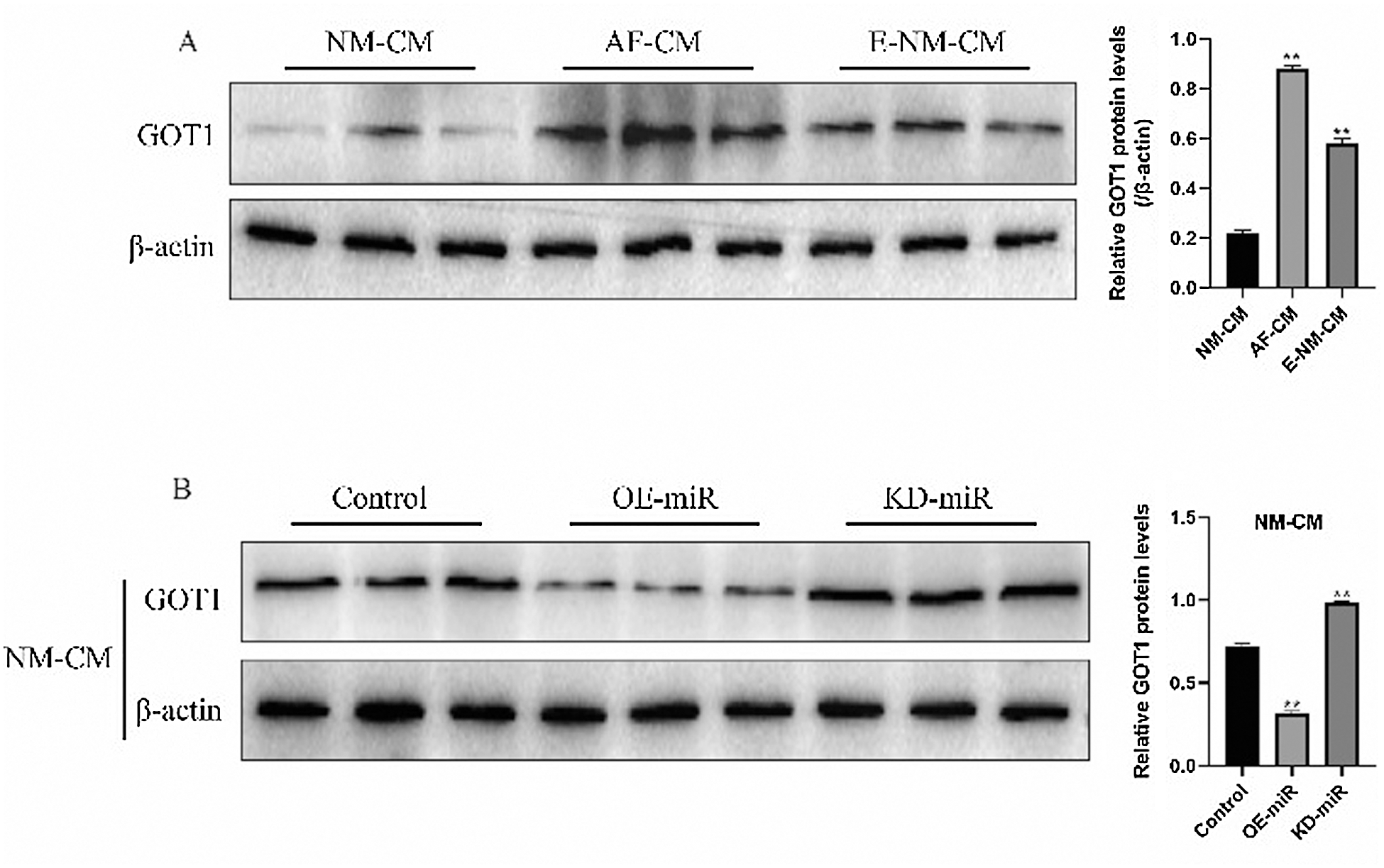
Figure S1: Western Blot was used to detect the expression levels of glutamic-oxaloacetic transaminase 1 in normal cardiomyocytes, atrial fibrillation cardiomyocytes, and Erastin-treated normal cardiomyocytes, respectively.
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |