DOI:10.32604/biocell.2021.013293
www.techscience.com/journal/biocell
| Biocell DOI:10.32604/biocell.2021.013293 | www.techscience.com/journal/biocell |
| Article |
APEX1 protects against oxidative damage-induced cardiomyocyte apoptosis
1Department of Cardiology, Tongji University Affiliated Tongji Hospital, Shanghai, 200065, China
2Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, Shanghai, 200032, China
3The West Coast New Area of Qingdao Traditional Chinese Medicine Hospital, Qingdao, 266500, China
4Cardiovascular Department of Internal Medicine, Central Hospital of Karamay, Karamay, 834000, China
*Address correspondence to: Xiaohong Liu, lxh51666@sohu.com; Zhiwen Ding, zhiwen_d@fudan.edu.cn
Received: 01 August 2020; Accepted: 23 October 2020
Abstract: Apurine/pyrimidine-free endonuclease 1 (APEX1) is a multifunctional enzyme that contributes to oxidization-mediated DNA-cleaved base excision repair and redox activation of transcription factors. However, the role of APEX1 during cardiomyocyte oxidative stress injury is not completely understood. In the present study, whether APEX1 protects oxidative damage-induced cardiomyocytes was investigated. mRNA and protein expression levels of APEX1 were downregulated in the mouse model of cardiac ischemia-reperfusion injury. Furthermore, the expression of APEX1 in hydrogen peroxide (H2O2)-treated neonatal mice cardiomyocytes was also decreased. APEX1 knockdown aggravated H2O2-treated cardiomyocyte apoptosis indexes. By contrast, APEX1 overexpression reversed H2O2-induced oxidative damage, as demonstrated by decreased caspase 3 and Bax expression levels. Moreover, homeobox A5 upregulated APEX1. The results of the present study indicated that APEX1 displayed protective effects against oxidative damage, suggesting that APEX1 may serve as a unique protective strategy for cardiac ischemia-reperfusion injury.
Keywords: Apurine/pyrimidine-free endonuclease 1; Cardiomyocyte apoptosis; Cardiac ischemia-reperfusion
It has been suggested that oxidative stress serves as a second messenger in a number of physiological and pathological processes, including cardiac ischemia-reperfusion (I/R) injury, diabetes, inflammation, atherosclerosis, and Alzheimer’s disease (Yodoi et al., 2001; Martindale and Holbrook, 2002; Calabrese et al., 2005). In physiological functions, the redox state of the cell is determined by the precise balance between reactive oxygen species (ROS) levels and endogenous antioxidant levels. However, intracellular ROS excessively accumulate during oxidative stress, which induces endogenous DNA damage, leading to cell apoptosis (Levonen et al., 2014; Espinosa-Diez et al., 2015; Di Marzo et al., 2018).
Apurine/pyrimidine-free endonuclease 1 (APEX1) consists of a large, conserved apurinic or apyrimidinic (AP) endonuclease domain and a unique N-terminal ref1 domain (Li and Wilson, 2014; Whitaker and Freudenthal, 2018). ROS can produce AP sites in DNA (Laev et al., 2017). APEX1 recognizes the AP site and cleaves the phosphodiester backbone 5’ to the lesion, resulting in DNA single-strand breaks (Dyrkheeva et al., 2016; Kuznetsova et al., 2018; Kladova et al., 2018). APEX1 not only directly participates in the DNA repair process but also regulates transcription by activating/modulating important transcription factors. For example, APEX1 activates/regulates critical transcription factors, including AP-1, early growth response 1, hypoxia-inducible factor 1 subunit-α, p53, and NF-κβ to influence biological events such as the stress response and inflammation (Schindl et al., 2001; Ando et al., 2008; Luo et al., 2008; Fantini et al., 2008; Poletto et al., 2016). However, few studies have detected the role of APEX1 in cardiac I/R injury.
The aim of the present study was to investigate the effects of APEX1 on cardiomyocyte oxidative stress injury. The results indicated that APEX1 expression was reduced by oxidative stress, and APEX1 effectively inhibited hydrogen peroxide (H2O2)-induced cell death by negatively regulating Bax and caspase 3.
Cell cultures and treatments of cardiomyocytes
Cardiomyocytes were obtained as previously described (Vidyasekar et al., 2015). Briefly, the hearts of 1–2 days old neonatal C57BL/6 mice were dissected, and the ventricles were minced and dissociated using 0.125% trypsin. Cells were cultured in gelatin-coated dishes with DMEM/F12 (Thermo Fisher Scientific Inc. catalog no. 11320033) supplemented with 10% FBS (Thermo Fisher Scientific Inc. catalog no. 10099133C). After 1.5 h, non-adherent cells were collected, and 106 cells were seeded into one well of collagen-coated silicon-based 6-well culture plates. The culture medium was changed to serum-free DMEM/F12 at 48 h. After 24 h, cardiomyocytes were treated with 10−4 M H2O2 (Matsuda et al., 2016).
The animals used in the I/R model were twenty SPF-level C57BL/6 male mice, 12 weeks old and weighing 25–30 g. They were purchased from GemPharmatech Co., Ltd., China. They were housed at 24 ± 2°C and 12 h–12 h dark–light cycles. Mice were randomly divided into sham group and I/R group. Both groups were anesthetized by intraperitoneal injection of a mixture of methylthiazine (10 mg/kg) and ketamine (150 mg/kg), tracheal intubation and ventilation (type 7025, Harvard Apparatus, March-Hugstetten, Germany). The left coronary artery was ligated for 30 min and perfused for 24 h to induce ischemia-reperfusion injury in the I/R group. All protocols were approved by the guidelines of the Animal Care and Use Committee of Fudan University and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
The three selected mouse APEX1-targeted shRNA sequences were obtained from BLOCK-iTTM RNAi Designer, Thermo Fisher Scientific, Inc.: shAPEX1-1, GCAAATCTGCCACACTCAAGA; shAPEX1-2, TCGGTATTCCAGTCTTACCAG; and shAPEX1-3, ATTGAGATCCCCACATAGCAC. The shRNA sequences were cloned into pLKO vector, and lentiviral particles were generated using 293T cells. pLKO.1-GFP lentiviral particles were used as a control.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from cultured cardiomyocytes using TRIzol® reagent. Total RNA was reverse transcribed into cDNA using the ReverTra Ace-α RT-PCR kit (Toyobo Life Science), according to the manufacturer’s protocol. Subsequently, qPCR was performed using the Bio-Rad IQ5 multicolor detection system (Bio-Rad Laboratories, Inc.) with Power SYBR Green PCR Master Mix (Takara Bio, Inc.) (2 min at 95°C for enzyme activation followed by 40 cycles of 15 s at 95°C and 30 s at 60°C for the amplification step). The mRNA expression levels were quantified using the 2−∆∆Ct method and normalized to the internal reference gene β-actin (Livak and Schmittgen, 2001).
Western blotting was performed according to standard protocols. Briefly, total protein was extracted from cardiomyocytes using RIPA buffer (Beyotime Institute of Biotechnology) and quantified using the bicinchoninic acid assay (Thermo Fisher Scientific Inc.). Proteins were separated via SDS-PAGE and transferred to PVDF membranes. Subsequently, the membranes were incubated with APEX1 (1:10000, catalog no. ab92744, Abcam), caspase 3 (1:1000, catalog no. 9664, Cell Signaling Technology), Bax (1:1000, catalog no. 2772, Cell Signaling Technology), HOXA5 (1:1000, catalog no. ab140636, Abcam). After washing three times, blotted membranes were then incubated with horseradish peroxidase-conjugated rabbit secondary antibody (1:5000, catalog no. KC-RB-035, Kang-Chen Biotechnology, Shanghai, China). β-actin (1:5000, catalog no. ab20272, Abcam) was used as the internal control.
The TUNEL assay was performed using the One-Step TUNEL Apoptosis assay kit (Beyotime Institute of Biotechnology). Cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100. Subsequently, cells were incubated with TUNEL test solution for 60 min and observed using a fluorescence microscope.
Data are presented as the mean ± SEM. Comparisons among multiple groups were analyzed using one-way ANOVA followed by the LSD post hoc test. Comparisons between two groups were analyzed using the Student’s t-test. P < 0.05 was considered to indicate a statistically significant difference. All experiments were repeated at least three times.
APEX1 is downregulated during heart I/R. To elucidate the role of APEX1 during heart I/R, a cardiac I/R model was established, and subsequently, ventricles were obtained. Compared with the control group, I/R significantly reduced APEX1 mRNA expression levels (Fig. 1A). In addition, the western blotting results indicated that I/R downregulated APEX1 protein expression (Fig. 1B). The results suggested that APEX1 served a role in I/R.
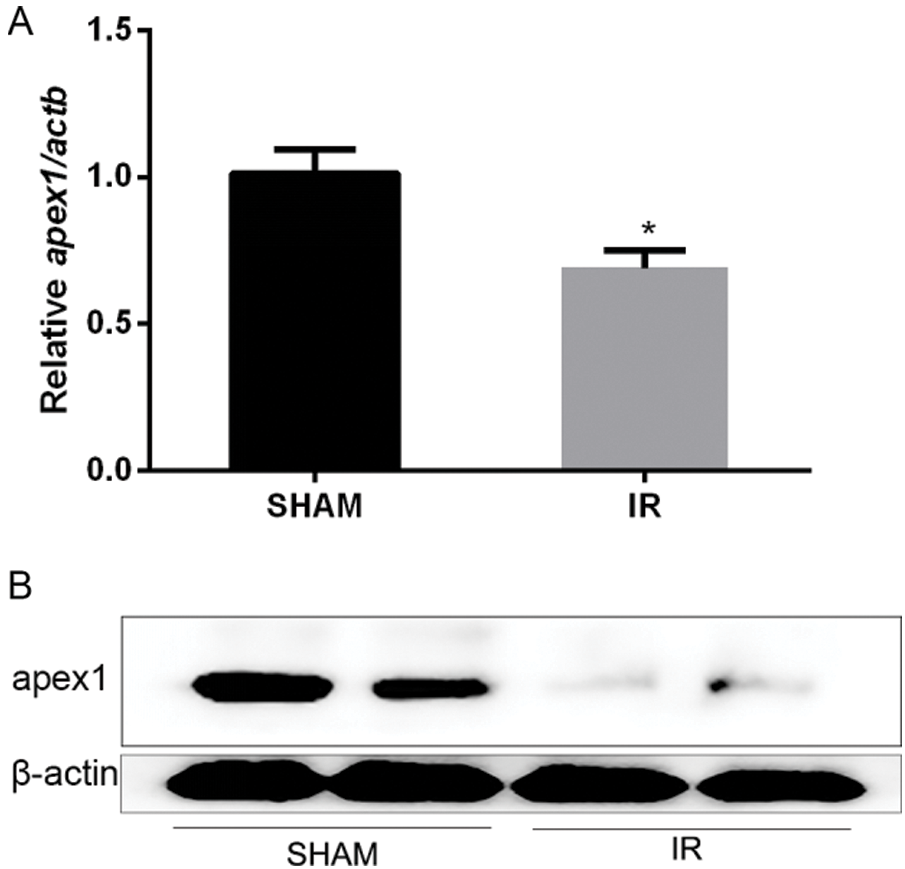
Figure 1: Relative expression of APEX1 in a mouse model of cardiac ischemia-reperfusion. APEX1 (A) mRNA and (B) protein expression levels. *P < 0.05 vs. control. APEX1, apurine/pyrimidine-free endonuclease 1.
APEX1 is downregulated in H2O2-treated cardiomyocytes. To investigate whether APEX1 contributed to the productive role against I/R in cardiomyocytes, cardiomyocytes were treated with H2O2. RT-qPCR and western blotting results suggested that H2O2 reduced APEX1 expression levels compared with the control group (Fig. 2). The results indicated that APEX1 displayed a protective role in cardiomyocytes.
Lentivirus-mediated APEX1 RNA interference vector construction. To investigate the protective role of APEX1 in cardiomyocytes, three shRNA sequences of APEX1 were selected to test their interference efficiency. The shRNA sequences were cloned into pLKO vector, and lentiviral particles were produced using the 293T cell line. RT-qPCR and western blotting were performed to detect the interference efficiency of the three candidate sequences. The RT-qPCR results indicated that the three candidate shRNAs knocked down APEX1 expression. APEX1 expression was reduced by 48%, 73%, and 30% by shAPEX1-1, shAPEX1-2, and shAPEX1-3, respectively (Fig. 3A). In addition, similar results were obtained by western blotting (Fig. 3B); therefore, shaAPEX1-2 was used for subsequent experiments.
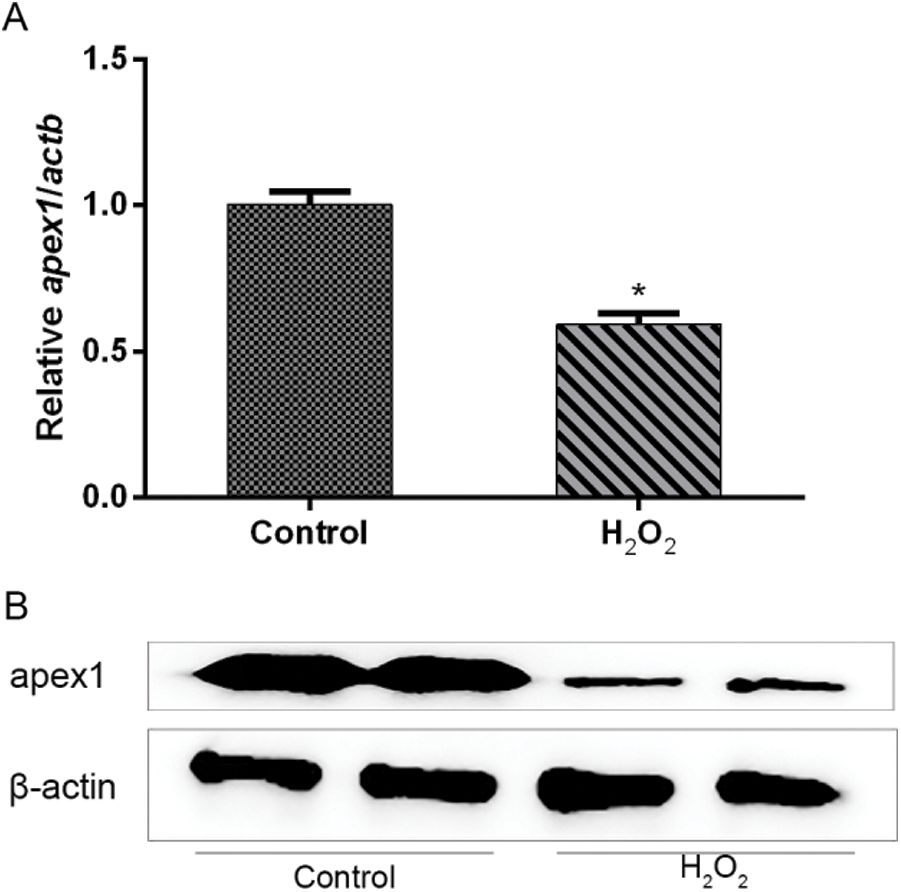
Figure 2: Relative expression of APEX1 in H2O2-treated cardiomyocytes. APEX1 (A) mRNA and (B) protein expression levels in cardiomyocytes following H2O2 (100 μM) treatment for 24 h. *P < 0.05 vs. control. APEX1, apurine/pyrimidine-free endonuclease 1.
APEX1 is associated with cell apoptosis in H2O2-treated cardiomyocytes. Subsequently, the protective effect of APEX1 against oxidative stress-induced damage of cardiomyocytes was investigated. H2O2 significantly increased Bax and caspase 3 expression levels compared with the control group. Additionally, APEX1 knockdown further enhanced the expression levels of Bax and caspase 3 (Fig. 4A). A similar increase in expression was observed using the TUNEL assay. Compared with the control group, H2O2 significantly induced TUNEL signaling, and APEX1 knockdown further increased the TUNEL signal (Fig. 4B). However, APEX1 overexpression reversed H2O2-induced Bax and caspase 3 expressions (Fig. 4C). The results suggested that APEX1 suppressed H2O2-induced cardiomyocyte apoptosis.
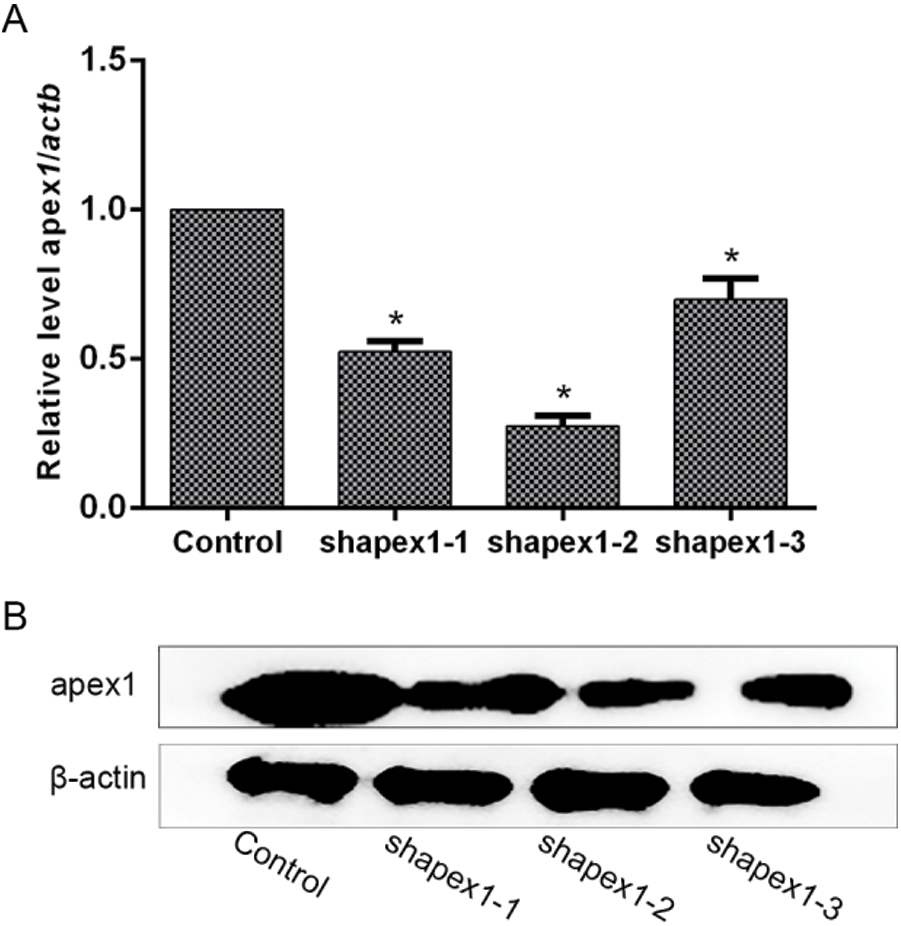
Figure 3: Candidates of shRNA-mediated APEX1 downregulation in cardiomyocytes. Transfection efficiency was determined by (A) reverse transcription-quantitative PCR and (B) western blotting. *P < 0.05 vs. Control. shRNA, short hairpin RNA; APEX1, apurine/pyrimidine-free endonuclease 1.
Homeobox A5 (HOXA5) is upstream of APEX1. Oxidative stress-induced cardiomyocyte apoptosis downregulated APEX1 expression. To identify which factors regulated APEX1 expression, the expression of several related transcription factors was knocked down using siRNAs. The results indicated that the HOXA5-targeted siRNA significantly decreased APEX1 expression levels (Fig. 5A). In addition, HOXA5 was downregulated in I/R injury (Fig. 5B). The results suggested that APEX1 was downstream of HOXA5.
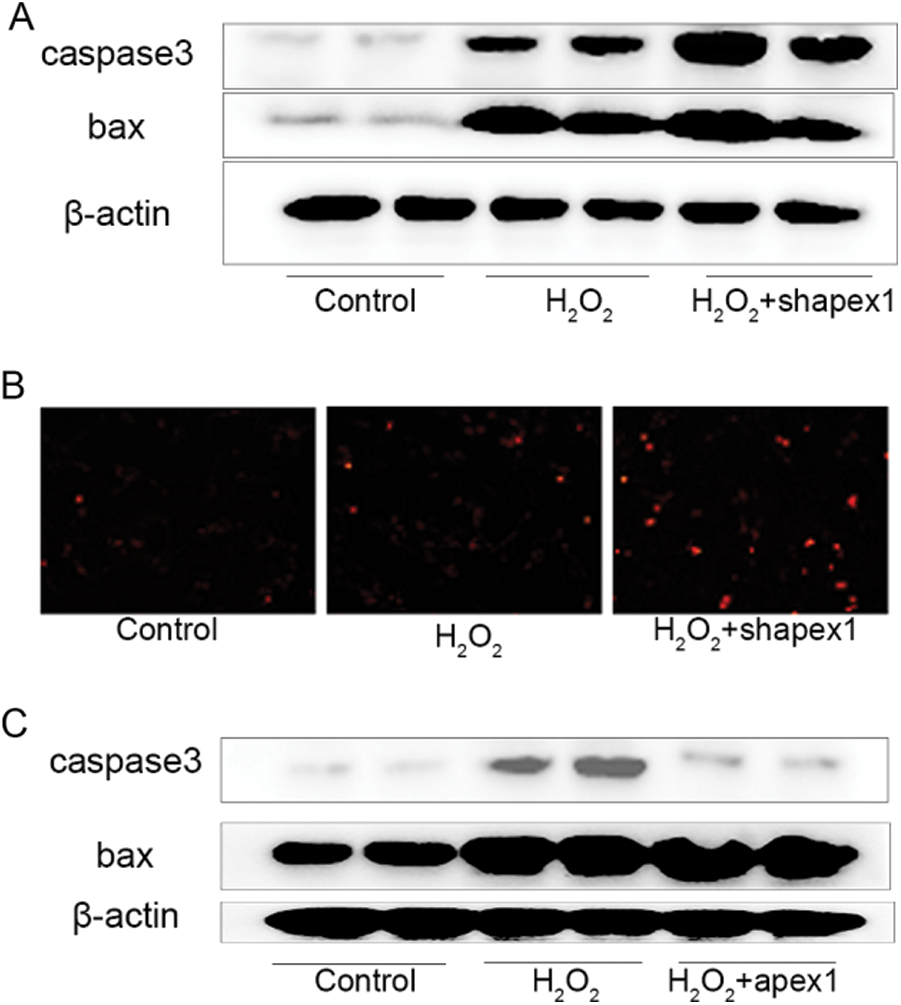
Figure 4: Role of APEX1 in H2O2. Caspase 3 and Bax protein expression levels were determined by western blotting following APEX1 knockdown in cardiomyocytes treated with H2O2 (100 μM) for 24 h. (B) The TUNEL assay was performed to assess the role of APEX1 in cardiomyocytes treated with H2O2 (100 μM) for 24 h. (C) Caspase 3 and Bax protein expression levels were determined by western blotting following APEX1 overexpression in cardiomyocytes treated with H2O2 (100 μM) for 24 h. APEX1, apurine/pyrimidine-free endonuclease 1.
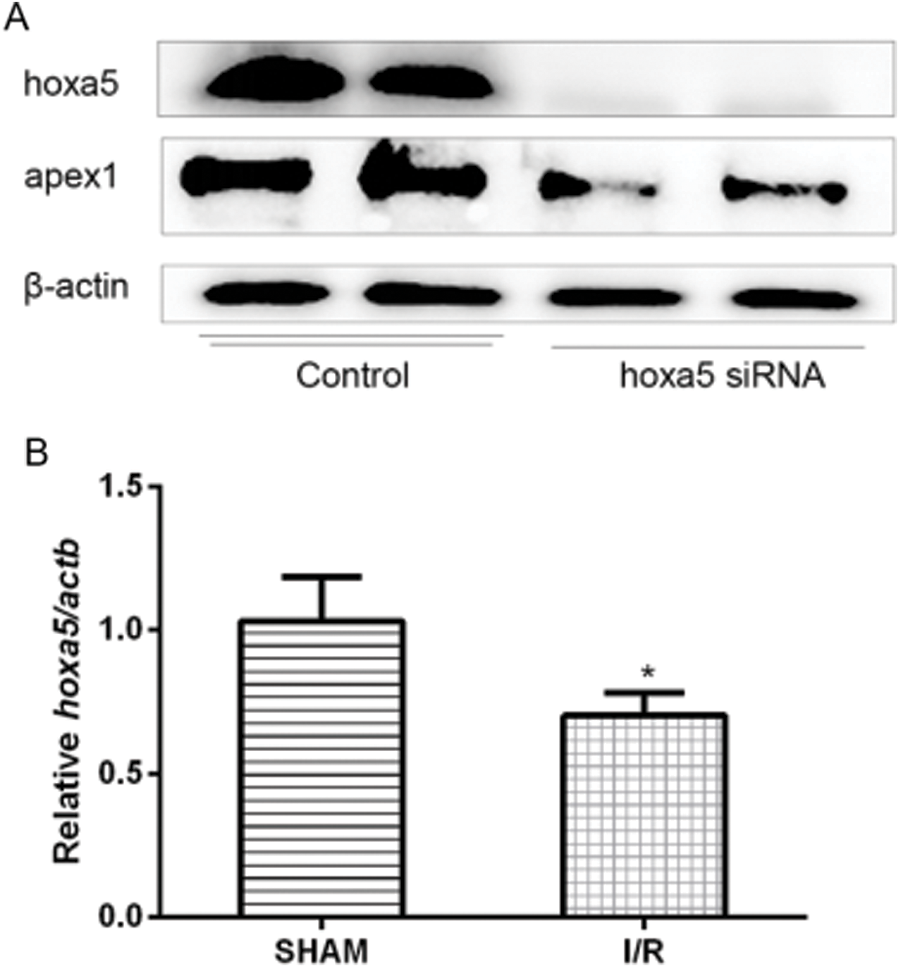
Figure 5: HOXA5 is upstream of APEX1. (A) APEX1 and HOXA5 protein expression levels were determined by western blotting in siRNA-transfected cardiomyocytes. (B) HOXA5 mRNA expression levels in a mouse model of I/R were determined by reverse transcription-quantitative PCR. *P < 0.05 vs. control. HOXA5, homeobox A5; APEX1, apurine/pyrimidine-free endonuclease 1; siRNA, small interfering RNA; I/R, ischemia-reperfusion.
Although reperfusion therapy is a potential therapeutic strategy for ischemic heart disease, as it reduces myocardial damage and improves clinical outcomes, reperfusion after ischemia-induced ROS production can lead to DNA damage, restrict myocardial repair and result in cardiac dysfunction (Görge et al., 1991). Recent studies have demonstrated that APEX1 serves an important role in the regulation of oxidative DNA damage in ischemic injury. In the present study, the two main findings may aid in understanding the protective effects of APEX1 against oxidative damage. First, APEX1 expression was reduced in the mouse model of cardiac ischemia-reperfusion in vivo and in vitro. Second, APEX1 protected against H2O2-induced oxidative damage. Therefore, the present study suggested a promising therapeutic strategy for cardiac ischemia-reperfusion injury.
APEX1 is a vital multifunctional protein that displays a pleiotropic role in controlling cellular oxidative stress and promoting genomic stability (Li and Wilson, 2014). APEX1, the main apurinic/apyrimidinic endonuclease in eukaryotic cells, serves a central role in the DNA base excision repair pathway, repairing uracil, alkylation, oxidation, and abasic sites, as well as DNA single-strand breaks (Dyrkheeva et al., 2016).
APEX1 has an important role in myocardial disease. Jeon et al. (2004) reported that heterozygous APEX1 mice displayed impaired endothelium-dependent vasodilation, reduced blood nitric oxide levels, and hypertension. Martinet et al. (2002) demonstrated that APEX1 expression was increased in human carotid atherosclerotic plaques. Furthermore, Jin et al. (2017) reported that serum APEX1 could be used to identify myocardial injury in viral myocarditis without an endomyocardial biopsy. However, the relationship between APEX1 and I/R has not been previously reported. The results of the present study suggested that APEX1 inhibited apoptosis during I/R, which indicated that APEX1 might serve as a potential therapeutic target for cardiac ischemia-reperfusion injury.
Our study is the first description of APEX1’s role in the heart. APEX1 expression was reduced by oxidative stress, which was regulated by HOXA5. APEX1 effectively inhibited hydrogen peroxide-induced cell death by negatively regulating Bax and caspase 3. The results of the present study indicated that APEX1 displayed protective effects against oxidative damage, suggesting that APEX1 may serve as a unique protective strategy for cardiac ischemia-reperfusion injury.
Availability of Data and Materials: The datasets used during the current study are available from the corresponding author on reasonable request.
Funding Statement: The present study was supported by the National Natural Science Foundation of China (Grant No. 81900245 and 81770395).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
Ando K, Hirao S, Kabe Y, Ogura Y, Sato I, Yamaguchi Y, Wada T, Handa H. (2008). A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Research 36: 4327–4336. DOI 10.1093/nar/gkn416. [Google Scholar] [CrossRef]
Calabrese V, Lodi R, Tonon C, D’Agata V, Sapienza M, Scapagnini G, Mangiameli A, Pennisi G, Stella AM, Butterfield DA. (2005). Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. Journal of the Neurological Sciences 233: 145–162. DOI 10.1016/j.jns.2005.03.012. [Google Scholar] [CrossRef]
Di Marzo N, Chisci E, Giovannoni R. (2018). The role of hydrogen peroxide in redox-dependent signaling: Homeostatic and pathological responses in mammalian cells. Cells 7: 156. DOI 10.3390/cells7100156. [Google Scholar] [CrossRef]
Dyrkheeva NS, Lebedeva NA, Lavrik OI. (2016). AP endonuclease 1 as a key enzyme in repair of apurinic/apyrimidinic sites. Biochemistry 81: 951–967. [Google Scholar]
Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, Lamas S. (2015). Antioxidant responses and cellular adjustments to oxidative stress. Redox Biology 6: 183–197. DOI 10.1016/j.redox.2015.07.008. [Google Scholar] [CrossRef]
Fantini D, Vascotto C, Deganuto M, Bivi N, Gustincich S, Marcon G, Quadrifoglio F, Damante G, Bhakat KK, Mitra S, Tell G. (2008). APE1/Ref-1 regulates PTEN expression mediated by Egr-1. Free Radical Research 42: 20–29. DOI 10.1080/10715760701765616. [Google Scholar] [CrossRef]
Görge G, Chatelain P, Schaper J, Lerch R. (1991). Effect of increasing degrees of ischemic injury on myocardial oxidative metabolism early after reperfusion in isolated rat hearts. Circulation Research 68: 1681–1692. DOI 10.1161/01.RES.68.6.1681. [Google Scholar] [CrossRef]
Jeon BH, Gupta G, Park YC, Qi B, Haile A, Khanday FA, Liu YX, Kim JM, Ozaki M, White AR, Berkowitz DE, Irani K. (2004). Apurinic/apyrimidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circulation Research 95: 902–910. DOI 10.1161/01.RES.0000146947.84294.4c. [Google Scholar] [CrossRef]
Jin SA, Lim BK, Seo HJ, Kim SK, Ahn KT, Jeon BH, Jeong JO. (2017). Elevation of serum APE1/Ref-1 in experimental murine myocarditis. International Journal of Molecular Sciences 18: 2664. DOI 10.3390/ijms18122664. [Google Scholar] [CrossRef]
Kladova OA, Bazlekowa-Karaban M, Baconnais S, Piétrement O, Ishchenko AA, Matkarimov BT, Iakovlev DA, Vasenko A, Fedorova OS, Le Cam E, Tudek B, Kuznetsov NA, Saparbaev M. (2018). The role of the N-terminal domain of human apurinic/apyrimidinic endonuclease 1, APE1, in DNA glycosylase stimulation. DNA Repair 64: 10–25. DOI 10.1016/j.dnarep.2018.02.001. [Google Scholar] [CrossRef]
Kuznetsova AA, Matveeva AG, Milov AD, Vorobjev YN, Dzuba SA, Fedorova OS, Kuznetsov NA. (2018). Substrate specificity of human apurinic/apyrimidinic endonuclease APE1 in the nucleotide incision repair pathway. Nucleic Acids Research 46: 11454–11465. DOI 10.1093/nar/gky912. [Google Scholar] [CrossRef]
Laev SS, Salakhutdinov NF, Lavrik OI. (2017). Inhibitors of nuclease and redox activity of apurinic/apyrimidinic endonuclease 1/redox effector factor 1 (APE1/Ref-1). Bioorganic and Medicinal Chemistry Letters 25: 2531–2544. DOI 10.1016/j.bmc.2017.01.028. [Google Scholar] [CrossRef]
Levonen AL, Hill BG, Kansanen E, Zhang J, Darley-Usmar VM. (2014). Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radical Biology and Medicine 71: 196–207. DOI 10.1016/j.freeradbiomed.2014.03.025. [Google Scholar] [CrossRef]
Li M, Wilson DM. (2014). Human apurinic/apyrimidinic endonuclease 1. Antioxid Redox Signal 20: 678–707. DOI 10.1089/ars.2013.5492. [Google Scholar] [CrossRef]
Livak KJ, Schmittgen TD. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 25: 402–408. DOI 10.1006/meth.2001.1262. [Google Scholar] [CrossRef]
Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL 2nd, Borch RF, Qiao X, Georgiadis MM, Kelley MR. (2008). Role of the multifunctional DNA repair and redox signaling protein APE1/Ref-1 in cancer and endothelial cells: Small-molecule inhibition of the redox function of APE1. Antioxidants & Redox Signal 10: 1853–1867. DOI 10.1089/ars.2008.2120. [Google Scholar] [CrossRef]
Martindale JL, Holbrook NJ. (2002). Cellular response to oxidative stress: Signaling for suicide and survival. Journal of Cellular Physiology 192: 1–15. DOI 10.1002/jcp.10119. [Google Scholar] [CrossRef]
Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. (2002). Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 106: 927–932. DOI 10.1161/01.CIR.0000026393.47805.21. [Google Scholar] [CrossRef]
Matsuda T, Zhai P, Sciarretta S, Zhang Y, Jeong JI, Ikeda S, Park J, Hsu CP, Tian B, Pan D, Sadoshima J, Del Re DP. (2016). NF2 activates Hippo signaling and promotes ischemia/reperfusion injury in the heart. Circulation Research 119: 596–606. DOI 10.1161/CIRCRESAHA.116.308586. [Google Scholar] [CrossRef]
Poletto M, Legrand AJ, Fletcher SC, Dianov GL. (2016). p53 coordinates base excision repair to prevent genomic instability. Nucleic Acids Research 44: 3165–3175. DOI 10.1093/nar/gkw015. [Google Scholar] [CrossRef]
Schindl M, Oberhuber G, Pichlbauer EG, Obermair A, Birner P, Kelley MR. (2001). DNA repair-redox enzyme apurinic endonuclease in cervical cancer: Evaluation of redox control of HIF-1α and prognostic significance. International Journal of Oncology 19: 799–802. [Google Scholar]
Vidyasekar P, Shyamsunder P, Santhakumar R, Arun R, Verma RS. (2015). A simplified protocol for the isolation and culture of cardiomyocytes and progenitor cells from neonatal mouse ventricles. European Journal of Cell Biology 94: 444–452. DOI 10.1016/j.ejcb.2015.06.009. [Google Scholar] [CrossRef]
Whitaker AM, Freudenthal BD. (2018). APE1: A skilled nucleic acid surgeon. DNA Repair 71: 93–100. DOI 10.1016/j.dnarep.2018.08.012. [Google Scholar] [CrossRef]
Yodoi J, Masutani H, Nakamura H. (2001). Redox regulation by the human thioredoxin system. Biofactors 15: 107–111. DOI 10.1002/biof.5520150212. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |