2021 45(1): 109-117
DOI:10.32604/biocell.2021.012048
www.techscience.com/journal/biocell
| BIOCELL 2021 45(1): 109-117 DOI:10.32604/biocell.2021.012048 | www.techscience.com/journal/biocell |
The role of mTOR signaling pathway in regulating autophagy in liver injury of TX mice with Wilson’s disease
Anhui University of Chinese Medicine, Hefei, 230038, China
*Address correspondence to: Nan Cheng, azychengnan@163.com
Received: 12 June 2020; Accepted: 15 September 2020
#These authors contributed equally to this work
Abstract: Wilson disease (WD), known as hepatolenticular degeneration (HLD), is a treatable autosomal recessive disorder of copper metabolism. Because copper deposits in the liver first, the liver is not only the original defective organ but also the most affected organ. The liver injury is also one of the main causes of death throughout the course of the disease. Therefore, the treatment of liver injury is the main task of WD treatment, which is of great significance to improve the prognosis of patients. Autophagy is a process that promotes cell survival through degradation, recycling, and absorption in order to maintain the normal physiological function of cells, while excessive autophagy can aggravate cell death. In view of the abnormal damage of liver cells in patients with WD, which may be related to the change of autophagy level, in this study, we established an animal model of WD through toxic milk (TX) mice, observed the change of autophagy level in the liver, and observed the change of liver damage in mice after treatment with autophagy inhibitors. It was found that the mTOR signaling pathway was activated and autophagy was inhibited in Wilson mouse liver. After treatment with rapamycin, the autophagy level of mice liver was upregulated, and the copper content of mice liver was reduced, and the damage was alleviated. TX mouse hepatocytes were isolated, after using siRNA to interfere with mTOR expression, the copper accumulation was significantly reduced, which was the same with RAPA treatment. The results showed that in TX mice, the damage caused by copper accumulation in the liver may be related to the decrease of autophagy level caused by the activation of the mTOR signaling pathway. Our findings suggested that RAPA or the use of siRNA targeting mTOR may have potential applications in the treatment of Wilson’s disease.
Keywords: Wilson disease (WD); TX mice; Autophagy; mTOR signaling pathway
Abbreviations
| AMPK | amp activated protein kinase |
| CP | ceruloplasmin |
| DMT | divalent metal transporter |
| hCTR1 | human copper transporter 1 |
| HLD | hepatolenticular degeneration |
| mTORC1 | rapamycin target protein complex 1 |
| PCA | penicillamine |
| SD | standard deviation |
| TGN | trans Golgi network |
| TX | toxic milk |
| WD | Wilson disease |
Wilson’s disease (WD), also known as hepatolenticular degeneration, is an autosomal recessive genetic disease. The mutation of the ATP7B gene in the 13q14.3 region of the chromosome caused the disorder of copper transport in hepatocytes (Czlonkowska et al., 2018), resulting in a large amount of copper deposition in the liver, brain, kidney, and other tissues, which may lead to liver cirrhosis, neurological/psychiatric symptoms, renal damage, corneal K-F ring, and other clinical manifestations, even life-threatening if serious (Dong et al., 2016; Dzieżyc-Jaworska et al., 2019). Copper is involved in many physiological processes such as mitochondrial respiratory chain, neurotransmitter synthesis, and iron metabolism. Excessive accumulation of copper leads to degeneration and necrosis of hepatocytes and neurons in the cerebral cortex (Czlonkowska et al., 1973). The accumulation of excessive copper in the liver and brain can also lead to apoptosis through oxidative stress and inflammation (Gupta, 2014). Maintaining copper homeostasis is very important for the normal life activities of the body, so the content of copper in tissues and cells must be controlled (Ferenci et al., 2015).
At present, the metal complexing agent, represented by penicillamine (PCA), is the main treatment for the disease. PCA has the advantages of high copper excretion in urine and significant improvement of symptoms, but it has many adverse reactions; about 30% of patients are forced to stop taking medicine because of serious adverse reactions (Aggarwal and Bhatt, 2018). Zinc sulfate or zinc gluconate can prevent the absorption of copper ions in the gastrointestinal tract and have less adverse reactions, so they are also used in the treatment of the disease. However, the effect of zinc on copper excretion is weak, and its effect on patients with obvious symptoms is poor. At present, zinc is mostly used in patients with early symptoms or maintenance treatment (Gromadzka et al., 2014).
The exact mechanism of abnormal copper metabolism in WD is not clear. Copper ions are mainly transported into intestinal mucosal cells by carriers such as human copper transporter 1 (hCTR1) (Gray et al., 2012) and divalent metal transporter (DMT1) (Przybylkowski et al., 2013) on the brush-like membrane of intestinal epithelial cells, and then transported from intestinal mucosal cells to the portal vein blood circulation by copper transporter ATP7A (Chun et al., 2017). In hepatocytes, the copper transport ATP7b located in the trans-Golgi network (TGN) combines the copper ions with the pre ceruloplasmin to form ceruloplasmin (CP), which is released into the blood and circulates to the whole body and is taken and utilized by other tissues and cells. If there is too much copper in hepatocytes, it will be excreted with bile under the action of ATP7b located on the side of the bile duct membrane (Sharma et al., 2018). Because of ATP7B gene mutation in WD patients, they have CP synthesis disorder and copper excretion disorder in bile and lead to excessive copper deposition in the liver and other organs.
Autophagy is a process that promotes cell survival through degradation, recycling, and absorption, so as to maintain the normal physiological functions of cells. However, excessive autophagy can aggravate cell death. In higher eukaryotic cells, autophagy is related to cell differentiation, development, and cell death, as well as various human diseases, such as cancer and neurodegenerative diseases (Antunes et al., 2018). The expression of autophagy-related genes changed in ATP7B deficient cells (Weiskirchen et al., 2019). In HepG2 cells without the ATP7B gene, inhibition of autophagy by ATG7 and ATG13 inhibitors could accelerate cell death (Polishchuk et al., 2019). Activation of autophagy in liver cells of WD patients could reduce apoptosis induced by copper ions (Polishchuk et al., 2019). These suggested that promoting autophagy could prevent copper-induced hepatocyte death and may play a protective role in WD. Autophagy is regulated by a variety of autophagy genes and proteins in the process of autophagy formation. It is upregulated by AMP-activated protein kinase (AMPK) and downregulated by rapamycin target protein complex 1 (mTORC1) (Di Nardo et al., 2014). PI3K-Akt-mTOR signaling pathway plays an important role in autophagy regulation.
In view of the copper accumulation and damage of liver cells in Wilson’s disease, in this study, we established an animal model of WD through TX mice (Medici and Huster, 2017), observed the change of autophagy level in the liver, the liver damage and copper accumulation in mice after treatment with autophagic agonist RAPA. We found that in TX mice, the damage caused by copper accumulation in the liver may be related to the decrease of autophagy level caused by the activation of the mTOR signaling pathway. RAPA or the use of siRNA targeting mTOR may have potential applications in the treatment of Wilson’s disease.
Toxic milk (TX) mice were purchased from the Jackson Laboratory in the United States. They were raised in Anhui experimental animal center. All animal experiments were conducted according to the Principles of Laboratory Animal Care (National Society for Medical Research). These experiments were approved by the Ethics Committee of Anhui University of traditional Chinese medicine.
The animal model was established according to the previous reports (Polishchuk et al., 2019). Six 8-week-old female TX mice (20 ± 2 g) (Wilson), and six 8-week-old homologous DL mouse (control), were used. The mice were executed, and the livers were taken out. Some of them were fixed with 4% paraformaldehyde and used for HE-staining. Some of them were extracted RNA and protein, respectively, after preparation of tissue homogenate to detect the expression of autophagy-related factors and copper content.
To further verify the role of autophagy in Wilson’s disease, a total of 18 female TX mice (8–10 week old, 20 ± 2 g) were randomly divided into 3 groups: Control group, Rapamycin (MedChemExpress, HY-10219) low-dose group (1.0 mg/kg) and RAPA high-dose group (2.0 mg/kg), they were injected intraperitoneally every other day. In the control group, saline of equal volume was injected into the abdominal cavity every day.
All mice were fed in isolated cages with independent air supply under the conditions of humidity (50–70%), 12 h light/dark cycle, and room temperature (18–22°C). Food and water were freely obtained for 8 weeks. Blood samples were collected from the venous plexus of mouse fundus to detect the content of copper in serum. The mice were killed by pulling the neck. The liver was divided into three parts: one was fixed with polyformaldehyde and used for pathological detection, the other was to detect the expression of autophagy-related factors, and the third was used for copper content detection.
The primary TX mouse hepatocytes were prepared according to the previous reports (Shi et al., 2005). The mice were executed and soaked in 75% alcohol for 10min. Opened the abdomen of the mouse along the abdomen, separated the inferior vena cava and portal vein, inserted the scalp needle into the inferior vena cava, and clamped the needle with the artery, perfused in EGTA buffer (5.4 mM KCl, 0.34 mM Na2HPO4, 0.44 mM KH2PO4, 0.5 mM EGTA, 0.14 mM NaCl, 25 mM Tricine) and the perfusion velocity was 10–15 mL/min at the beginning. After a few seconds, the liver turned yellow and the perfusion velocity changed to 15–20 mL/min. After 20 mL perfusion, EGTA was changed to 0.035% collagenase to perfusion, the perfusion velocity was 10–15 mL/min, the liver became pink and transparent after 40–50 mL perfusion, and a layer of water appeared on the surface of the liver. The sign of the end of perfusion was the local collapse of the liver, which was difficult to recover after pressing. After the perfusion, carefully cut off the liver, cut it into pieces in a 60-mm-plate, added a proper amount of 0.019% collagenase, placed it in a 37°C water bath shaker at 50×g for 8 min. Filtered twice with 100 mesh stainless steel filter screen, transferred the filtrate to centrifuge tube (15 mL), and centrifuged at 4°C, 50×g for 3 min. The supernatant was removed, and the cells were re-suspended in a pre-cooled DMEM medium containing 10% FBS. The cells were centrifuged and washed twice with PBS. The cells were re-suspended with a medium containing insulin and dexamethasone and cultured at 37°C containing 5% CO2. siRNA targeting mTOR (Santa Cruz Biotechnology, sc-35410) was used to downregulate the expression of mTOR in mouse hepatocytes using Lipofectamine 3000 (Life Technologies, Carlsbad, CA, USA) according to the manufactured manual. The cells were divided into the WT control group, RAPA (10 nM) group, the siRNA control group, and the siRNA-MTOR group. The cells in the control group were cultured regularly, 10 nM of RAPA was added in the medium in RAPA (10 nM) group, siRNA-MTOR group, and siRNA control group were infected with mTOR siRNA and its control siRNA, respectively.
Determination of copper in liver tissue and serum
The mouse liver tissue (1 g) was placed in an oven at 80°C for 8–12 h and weighed it repeatedly until the weight is constant. Inductively coupled plasma mass spectrometer (Agilent 7500a ICP-MS, Agilent company, USA) was used to determine the content of copper in tissues. Serum samples were measured directly.
In primary mouse hepatocytes, 1 × 107 cells were heated in an oven at 80°C for 8–12 h, repeatedly weighed until the mass was constant. The content of copper in tissue was determined by inductively coupled plasma mass spectrometer (Agilent 7500a ICP-MS, Agilent company, USA). At the same time, the total protein was extracted from the same amount cells, BAC protein quantitative Kit was used to detect the protein content, and take the ratio of Cu/Protein content (ng/mg) as the intracellular copper content.
Detection of ceruloplasmin (CER) activity in serum
The activity of ceruloplasmin was detected by the p-phenylenediamine test kit (Nanjing Jiancheng Biology) according to the instructions.
HE-staining to detect the histopathological changes of liver
After dewaxing the paraffin liver section, HE stain was used to observe the degree of liver tissue damage, including morphology, structure, and distribution of hepatocytes.
Total RNA was extracted using the TRIzol kit according to the manufacturer’s instructions. Total RNA (1 μg) was subjected to reverse transcription using TaKaRa cDNA synthesis Kit. Real-time PCR was performed using SYBR Green PCR Master Mix. At the end of each reaction, a melting curve analysis was performed to confirm the absence of primer dimmers. GAPDH gene was used as an internal control for the normalization of RNA quantity and quality differences in all samples. Quantifications of target genes mRNA was performed using the2−ΔΔCt method. The thermocycler parameters were as follows: 95°C for 10 min, 40 cycles of 95°C for 20 s, 60°C for 30 s, and 72°C for 30 s. Primers’ sequences were listed in Tab. 1.
Table 1: Primers used in this study

Total protein was extracted from tissues and cells in different groups, and protein concentration was determined using BCA. Proteins (50 μg per lane) were separated using 12% SDS-PAGE. Proteins were then transferred to a PVDF membrane. The PVDF membrane was rinsed with TBS for 10–15 min, placed in TBS/T blocking buffer containing 5% (w/v) skimmed milk powder. It was incubated at 4°C overnight following the addition of an appropriate dilution of primary antibodies (p-Akt (s473), abcam, ab18206; Akt, abcam, ab8805; mTOR, abcam, ab109268; LC3; ATG7, abcam, ab52472; Beclin 1, abcam, ab207612; p62, abcam, ab56416; p-p70S6K, abcam, ab131436; p70S6K, abcam, ab32529; GAPDH, abcam, ab16891). The membrane was then rinsed with TBST three times and then incubated at room temperature for 1 h with horseradish peroxidase-labeled secondary antibody. The membrane was then rinsed three times with TBST. Protein bands were detected using an enhanced chemiluminescence kit (Perkin-Elmer, Inc.) and quantified as the ratio to GAPDH. Quantification was performed using Image J software.
The SPSS 22.0 software (SPSS Inc., Chicago, IL, USA) was used to carry out statistical analysis. All data are presented as the mean ± standard deviation (SD). The differences among groups were evaluated by Student’s t-test and one-way ANOVA. p < 0.05 was considered to be significant.
Copper accumulation and abnormal liver tissue structure in TX mouse model
Compared with that of the control group, the HE-staining results showed that the arrangement of hepatocytes in the control group was disordered. Most hepatocytes had extensive and severe vesicular degeneration and vacuolation (Fig. 1A). The content of copper ion in liver tissue increased significantly (Fig. 1B), the content of copper ion in serum decreased significantly, and the activity of ceruloplasmin increased significantly (Figs. 1C and 1D).
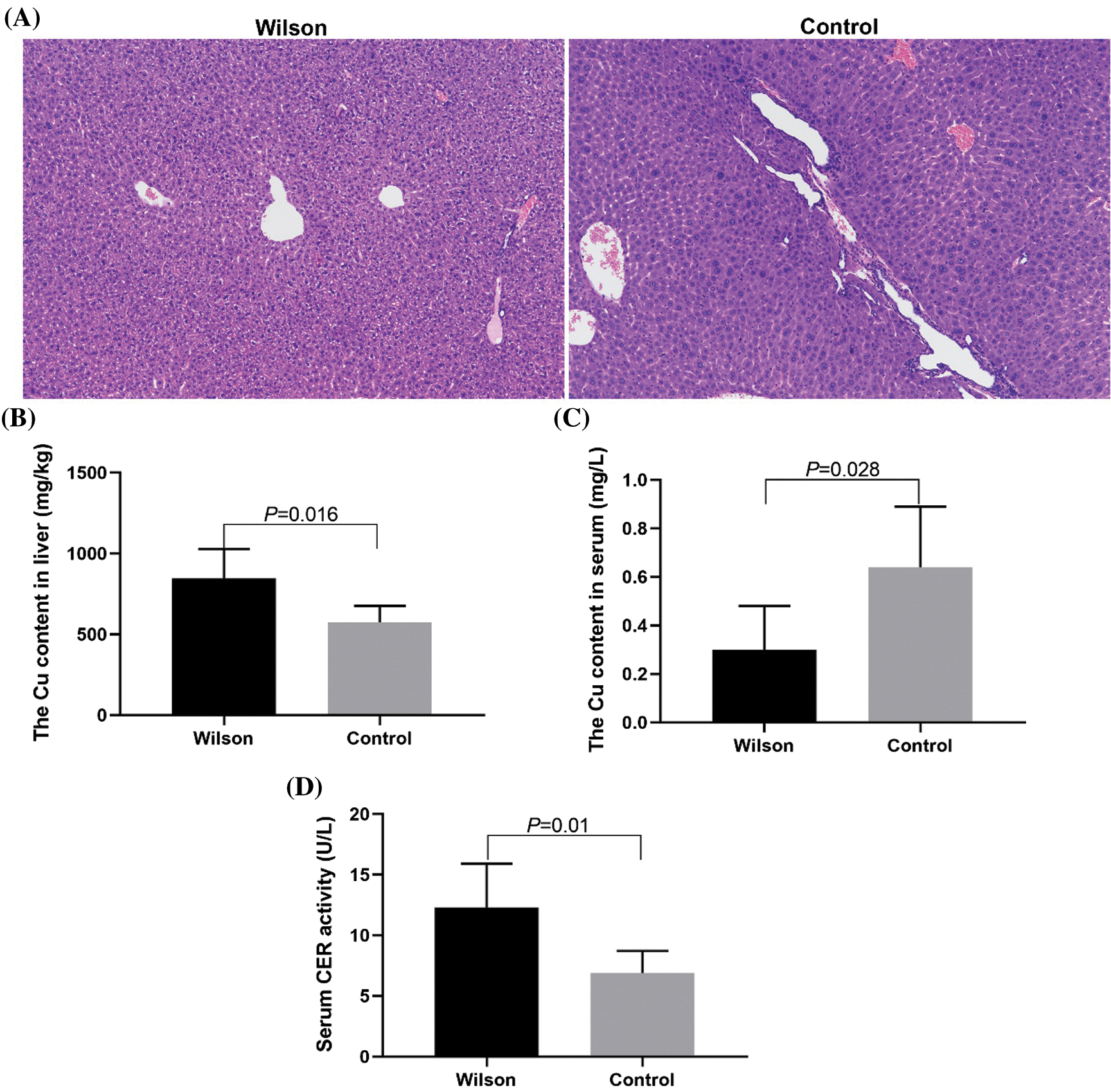
Figure 1: TX mice showed the characteristics of Wilson’s disease with liver injury and abnormal copper metabolism and could be treated by Penicillamine.
Activation of mTOR signaling pathway and inhibition of autophagy in the liver of TX mouse model
RT-PCR results showed that the expression levels of ATG7 and Beclin 1 mRNA decreased (Fig. 2A). Western blotting results showed that compared with the control group, p-Akt (s473) level and mTOR (s2448) level were increased, while the expression levels of LC3-II/LC3-1 ratio, ATG7, and Beclin 1 were decreased in TX mice (Figs. 2B–2E). These results suggested that the accumulation of copper in the liver of TX mice may be related to the activation of the mTOR signaling pathway and the inhibition of autophagy.
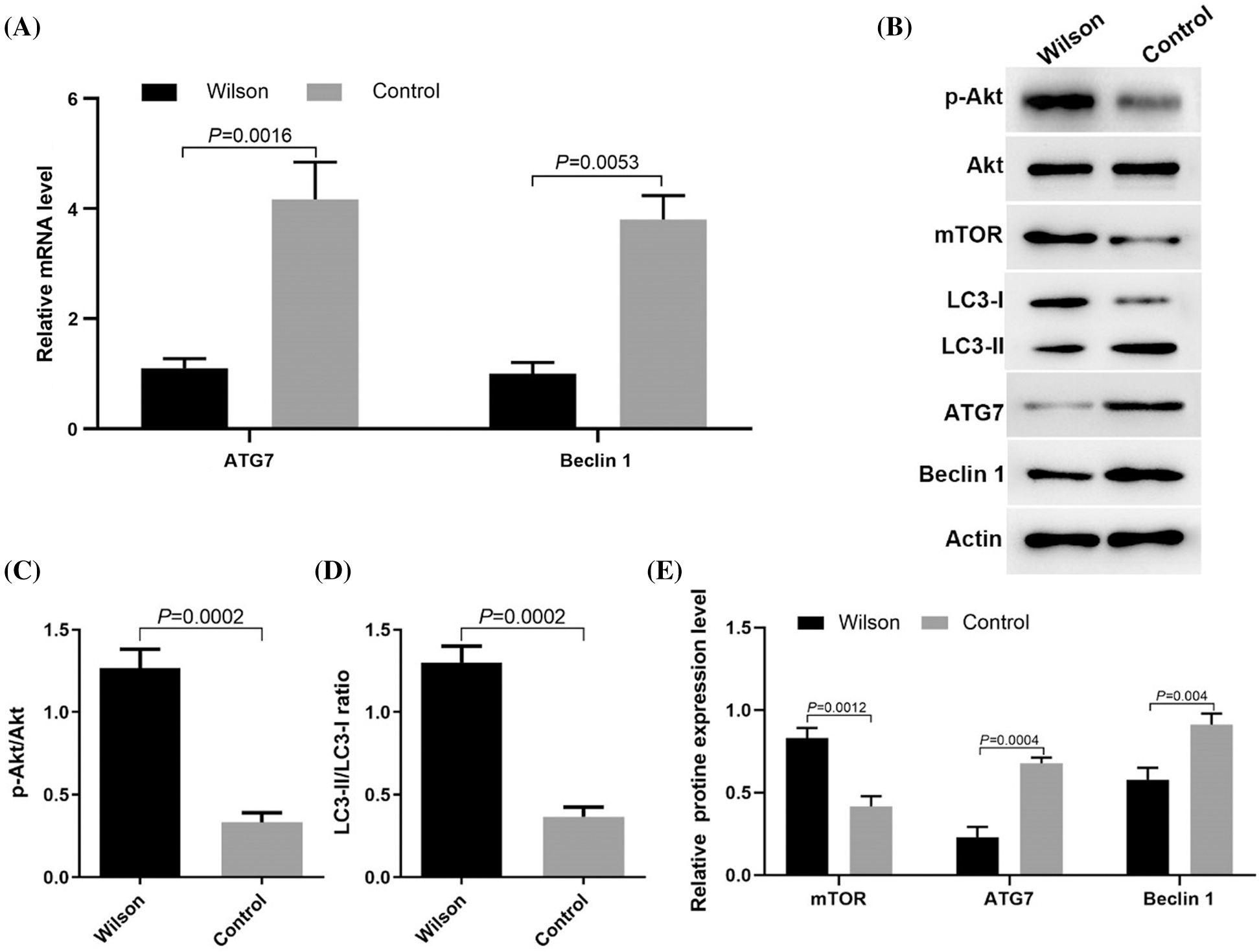
Figure 2: Copper accumulation in the liver of Wilson’s disease was related to the activation of the mTOR signaling pathway and the inhibition of autophagy.
RAPA could reduce liver injury and copper accumulation in TX mouse model
HE staining results showed that that the degree of liver damage and copper ion content in liver tissues in the RAPA low dose group and high dose group was lower than that in the control group, and the effect in the high dose group was more significant (Fig. 3A), the content of copper in the liver (Fig. 3B) and the activity of ceruloplasmin in serum (Fig. 3D) were significantly lower than that in the control group, while the serum copper content was significantly higher than that in the control group (Fig. 3C).
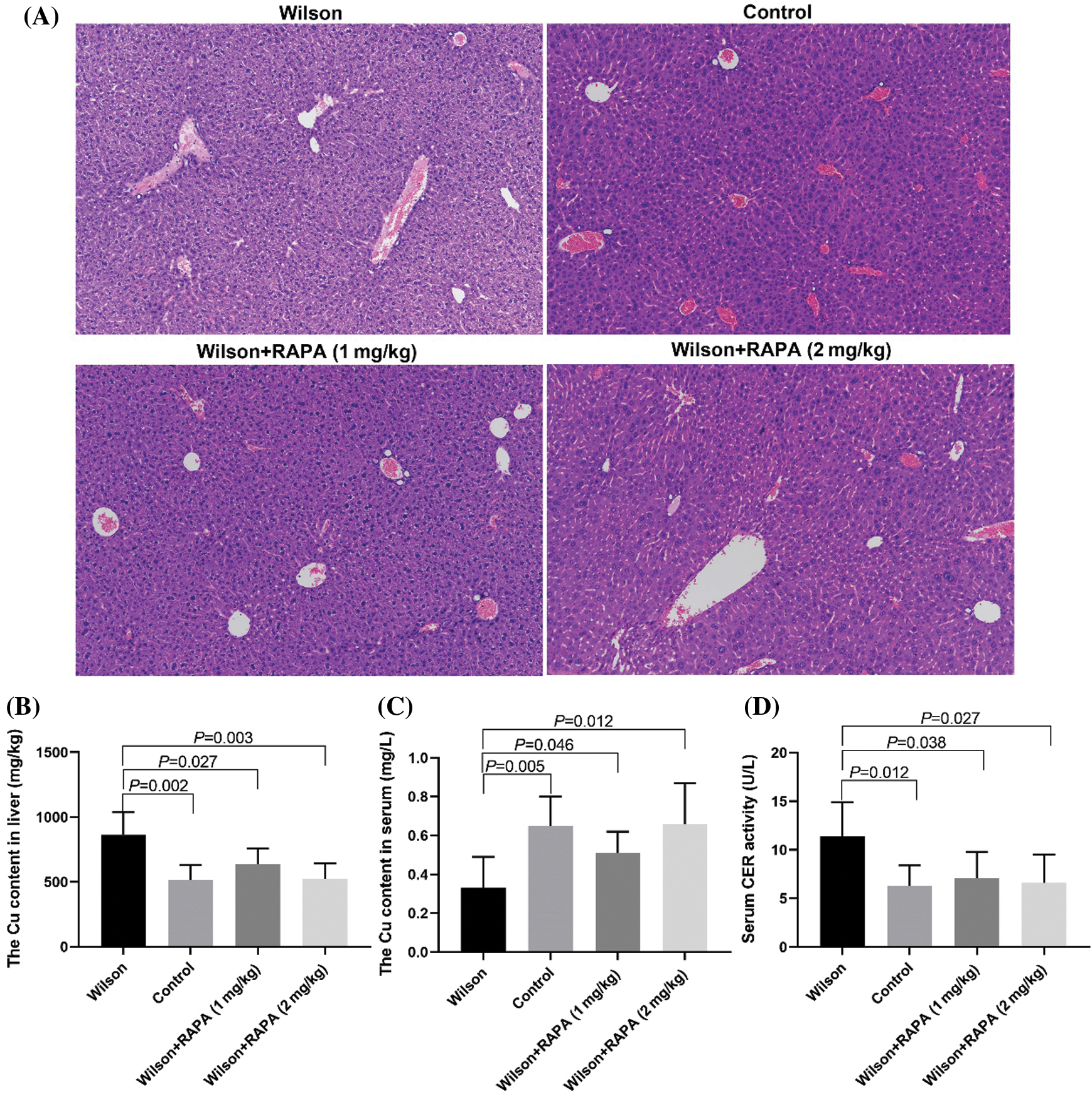
Figure 3: RAPA could reduce liver injury in TX mice.
RAPA promoted autophagy of hepatocytes in TX mouse model
RT-PCR results showed that RAPA promoted the expression levels of ATG7, Beclin 1, and p62 mRNA (Fig. 4A). Western blotting results showed RAPA promoted the expression levels of LC3-II/LC3-1 ratio, ATG7, and Beclin 1 and inhibited p62 expression in liver tissues of TX mice, especially in high dose group (Figs. 4B–4D). These results suggested that RAPA could reduce copper accumulation and damage in Wilson mouse liver by inhibiting the mTOR signaling pathway to promote autophagy.
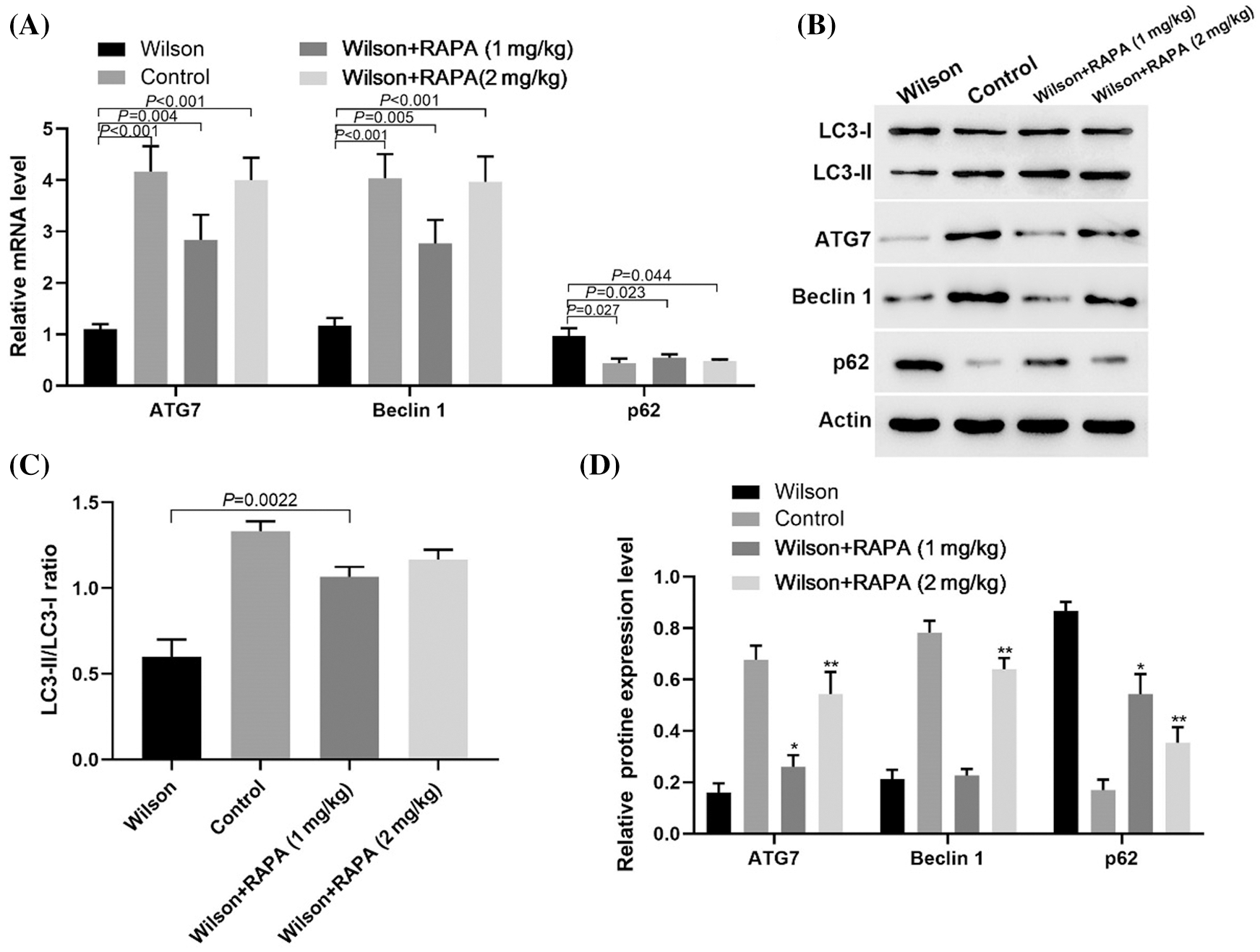
Figure 4: RAPA alleviated Wilson’s liver injury by promoting autophagic related molecules expression.
Down regulation of mTOR expression in TX mouse hepatocytes could reduce copper accumulation by promoting autophagy
In TX mouse hepatocytes, downregulation of mTOR expression by siRNA could promote copper ion efflux as well as RAPA treatment (Fig. 5A). Western blotting results showed that the expression levels of LC3-II/LC3-1 ratio, ATG7, and Beclin 1 expression increased after the downregulation of mTOR expression, while p-p70S6K and p62 expression decreased (Figs. 5B–5E). These results suggested that the mTOR signaling pathway increased copper accumulation in TX mouse hepatocytes by inhibiting autophagy.
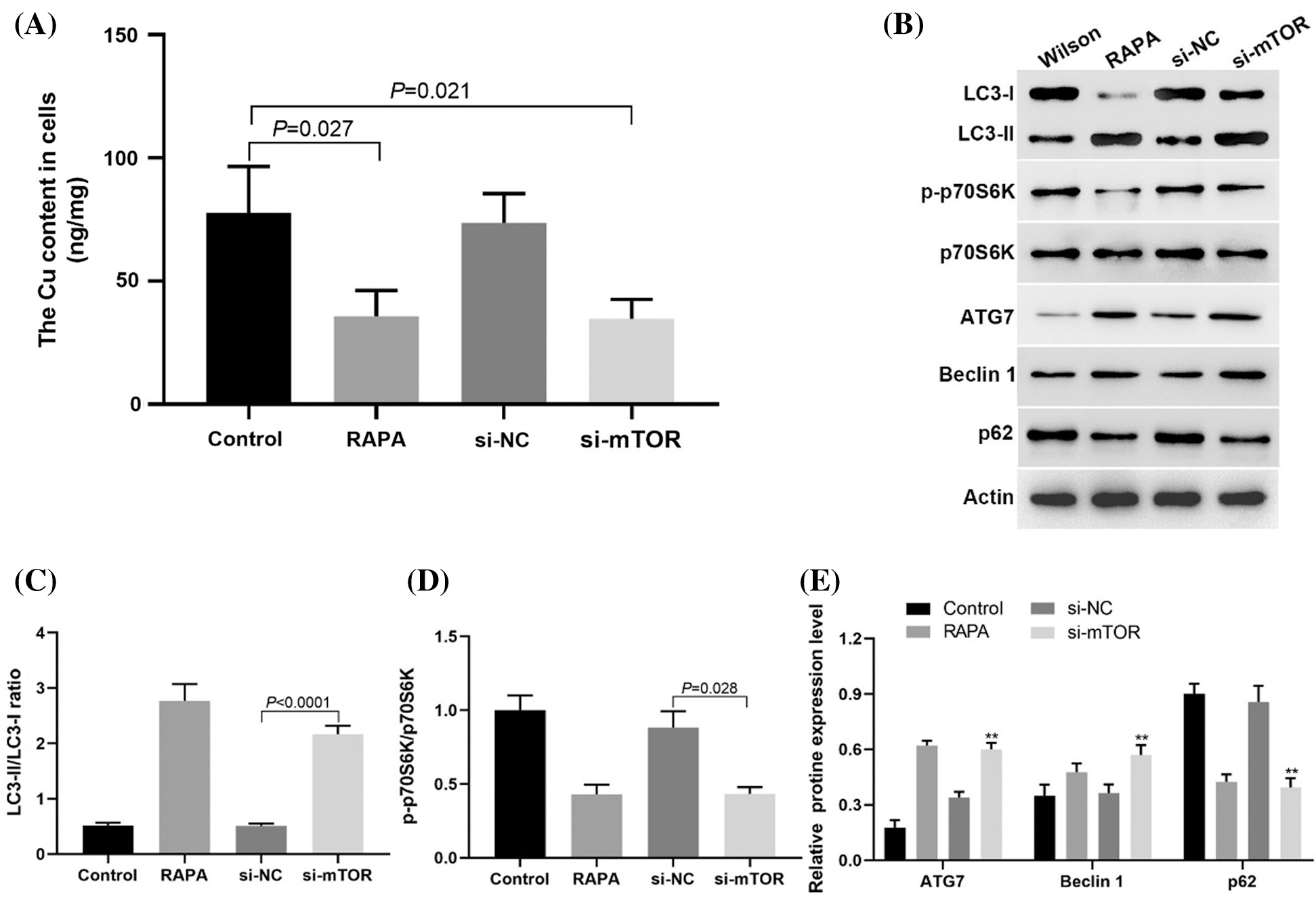
Figure 5: Downregulation of mTOR expression could inhibit the copper accumulation in liver cells of TX mice.
In this study, we found that in the WD model of TX mice, the damage caused by copper accumulation may be related to the activation of the mTOR signaling pathway and the decrease of autophagy level in liver cells. RAPA could promote the autophagy of liver cells of TX mice, reduce the accumulation of copper and liver injury by upregulating the ratio of LC3-II/LC3-1, the expression of ATG7 and Beclin 1, and inhibiting the accumulation of p62. In vitro experiments showed that the use of siRNA to down regulate the expression of mTOR could inhibit p-p70S6K expression and block mTOR signaling pathway, up regulate the ratio of LC3-II/LC3-1, the expression of ATG7 and Beclin 1, which was the same as that of RAPA treatment.
WD is the main cause of non-viral liver disease, accounting for 48.92% (Bandmann et al., 2015). The high copper state can lead to the changes and dysfunction of organelles and cell structures such as mitochondria, lysosomes, ribosomes, plasmalemma, microtubules, etc., and also affect the activity of many enzymes in cells and nuclear DNA. These changes can lead to complex pathological changes in organs and corresponding clinical symptoms (Fieten et al., 2016). Autophagy is an evolutionarily conserved and highly regulated steady-state process. Cytoplasmic macromolecules and organelles will be degraded through the removal and lysosomal system transformation. Autophagy plays an important role in cell growth, development, and homeostasis, and helps to maintain the balance between the synthesis, degradation, and subsequent circulation of cell products under normal physiological conditions (Parzych and Klionsky, 2014). Autophagy defects have been found to be related to neuronal loss in neurodegenerative diseases, in which abnormal proteins and damaged organelles cannot be removed from neurons (Guo et al., 2018). Autophagy induced and subsequent neuronal death occur in several pathological states of the nervous system, including ischemia, trauma, toxicity induced neurotoxicity, and neurodegenerative protein aggregation disease.
Abnormal autophagy was also found in other liver diseases. In nonalcoholic fatty liver disease, high levels of energy substrates (such as ATP), insulin, or free fatty acids have a negative regulatory effect on autophagy (Madrigal-Matute and Cuervo, 2016). Autophagy is important for the stability of the intracellular environment of hepatocytes because protein aggregates, lipid droplets, or organelles are eliminated through this pathway (Ueno and Komatsu, 2017). Autophagy inhibition is associated with the occurrence of spontaneous liver tumors (Poillet-Perez et al., 2018). Recent studies showed that autophagy protected hepatocytes from copper poisoning, and hepatocytes may be in a state of autophagy inhibition in Wilson’s disease liver (Ma et al., 2017; Bialik et al., 2018), which was consistent with the observation results of this study.
The molecular mechanism of autophagy regulation is a very complex biological process that involves many different signaling pathways. PI3K/Akt pathway is the central regulator of signal transduction pathway involved in cell growth, as well as the regulator of signal transduction pathway involved in cell growth, cell survival, and metabolism. More and more evidence showed that the PI3K/Akt pathway played a key role in regulating autophagic cell death (Li et al., 2019). Zheng et al. (2014) reported that melatonin inhibited autophagy induced by ischemia/reperfusion by enhancing the activation of the PI3K/Akt pathway. Inhibition of the PI3K/Akt pathway could lead to the autophagic death of cardiomyocytes (Hou et al., 2014).
Akt, also known as protein kinase B, is the main mediator of the PI3K/Akt signaling pathway. Akt is located downstream of the PI3-K signal pathway and is activated by phosphorylation. The mammalian target of rapamycin kinase (mTOR) is the main regulatory factor of autophagy and the downstream target of the PI3K/Akt pathway. Akt can activate another mTOR in cells. The inhibition of autophagy depends on the activation of the mTOR signal. Zhang et al. showed that lead-exposure induced autophagy and autophagic cell death in rat hippocampus by inhibiting Akt/mTOR signaling pathway (Meng et al., 2016). It was reported that silica nanoparticles induced autophagy of endothelial cells through the PI3K/Akt/mTOR signaling pathway (Duan et al., 2014). These studies showed that the PI3K/Akt/mTOR pathway inactivation promoted the upregulation of autophagy.
In conclusion, our results suggested that the mTOR signaling pathway was inhibited, leading to over autophagy, which was related to liver injury caused by copper accumulation in Wilson’s disease. Our findings may provide a new idea for the treatment of Wilson’s disease with anti-autophagy.
Availability of Data and Materials: The data used to support the findings of this study are available from the corresponding author upon request.
Funding Statement: This study was supported by Natural Science Foundation of Anhui Province (1908 085MH266) and National Natural Science Foundation of China (81673948).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding this study.
Aggarwal A, Bhatt M. (2018). Advances in treatment of Wilson disease. Tremor and Other Hyperkinetic Movements 8: 525. DOI 10.5334/tohm.435. [Google Scholar] [CrossRef]
Antunes F, Erustes AG, Costa AJ, Nascimento AC, Bincoletto C, Ureshino RP, Pereira GJS, Smaili SS. (2018). Autophagy and intermittent fasting: The connection for cancer therapy? Clinics 73: e814s. DOI 10.6061/clinics/2018/e814s. [Google Scholar] [CrossRef]
Bandmann O, Weiss KH, Kaler SG. (2015). Wilson’s disease and other neurological copper disorders. Lancet Neurology 14: 103–113. DOI 10.1016/S1474-4422(14)70190-5. [Google Scholar] [CrossRef]
Bialik S, Dasari SK, Kimchi A. (2018). Autophagy-dependent cell death–where, how and why a cell eats itself to death. Journal of Cell Science 131: jcs215152. DOI 10.1242/jcs.215152. [Google Scholar] [CrossRef]
Chun H, Catterton T, Kim H, Lee J, Kim BE. (2017). Organ-specific regulation of ATP7A abundance is coordinated with systemic copper homeostasis. Scientific Reports 7: 12001. DOI 10.1038/s41598-017-11961-z. [Google Scholar] [CrossRef]
Czlonkowska A, Galewicz A, Rodo M, Wehr H. (1973). Observations on copper metabolism in Wilson’s disease. Acta Universitatis Carolinae. Medica. Monographia 56: 175–177. [Google Scholar]
Czlonkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, Rybakowski JK, Weiss KH, Schilsky ML. (2018). Wilson disease. Nature Reviews Disease Primers 4: 21. DOI 10.1038/s41572-018-0018-3. [Google Scholar] [CrossRef]
Di Nardo A, Wertz MH, Kwiatkowski E, Tsai PT, Leech JD, Greene-Colozzi E, Goto J, Dilsiz P, Talos DM, Clish CB, Kwiatkowski DJ, Sahin M. (2014). Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Human Molecular Genetics 23: 3865–3874. DOI 10.1093/hmg/ddu101. [Google Scholar] [CrossRef]
Dong Y, Ni W, Chen WJ, Wan B, Zhao GX, Shi ZQ, Zhang Y, Wang N, Yu L, Xu JF, Wu ZY. (2016). Spectrum and classification of ATP7B variants in a large cohort of Chinese patients with Wilson’s disease guides genetic diagnosis. Theranostics 6: 638–649. DOI 10.7150/thno.14596. [Google Scholar] [CrossRef]
Duan J, Yu Y, Yu Y, Li Y, Wang J, Geng W, Jiang L, Li Q, Zhou X, Sun Z. (2014). Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. International Journal of Nanomedicine 9: 5131–5141. DOI 10.2147/IJN.S71074. [Google Scholar] [CrossRef]
Dzieżyc-Jaworska K, Litwin T, Członkowska A. (2019). Clinical manifestations of Wilson disease in organs other than the liver and brain. Annals of Translational Medicine 7: S62. DOI 10.21037/atm.2019.03.30. [Google Scholar] [CrossRef]
Ferenci P, Litwin T, Seniow J, Członkowska A. (2015). Encephalopathy in Wilson disease: Copper toxicity or liver failure? Journal of Clinical and Experimental Hepatology 5: S88–S95. DOI 10.1016/j.jceh.2014.09.002. [Google Scholar] [CrossRef]
Fieten H, Gill Y, Martin AJ, Concilli M, Dirksen K, van Steenbeek FG, Spee B, van den Ingh TS, Martens EC, Festa P, Chesi G, van de Sluis B, Houwen RH, Watson AL, Aulchenko YS, Hodgkinson VL, Zhu S, Petris MJ, Polishchuk RS, Leegwater PA, Rothuizen J. (2016). The Menkes and Wilson disease genes counteract in copper toxicosis in Labrador retrievers: A new canine model for copper-metabolism disorders. Disease Models & Mechanisms 9: 25–38. DOI 10.1242/dmm.020263. [Google Scholar] [CrossRef]
Gray LW, Peng F, Molloy SA, Pendyala VS, Muchenditsi A, Muzik O, Lee J, Kaplan JH, Lutsenko S. (2012). Urinary copper elevation in a mouse model of Wilson’s disease is a regulated process to specifically decrease the hepatic copper load. PLoS One 7: e38327. DOI 10.1371/journal.pone.0038327. [Google Scholar] [CrossRef]
Gromadzka G, Karpinska A, Przybylkowski A, Litwin T, Wierzchowska-Ciok A, Dziezyc K, Chabik G, Czlonkowska A. (2014). Treatment with D-penicillamine or zinc sulphate affects copper metabolism and improves but not normalizes antioxidant capacity parameters in Wilson disease. Biometals 27: 207–215. DOI 10.1007/s10534-014-9706-y. [Google Scholar] [CrossRef]
Guo F, Liu X, Cai H, Le W. (2018). Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathology 28: 3–13. DOI 10.1111/bpa.12545. [Google Scholar] [CrossRef]
Gupta S. (2014). Cell therapy to remove excess copper in Wilson’s disease. Annals of the New York Academy of Sciences 1315: 70–80. DOI 10.1111/nyas.12450. [Google Scholar] [CrossRef]
Hou X, Hu Z, Xu H, Xu J, Zhang S, Zhong Y, He X, Wang N. (2014). Advanced glycation endproducts trigger autophagy in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovascular Diabetology 13: 78. DOI 10.1186/1475-2840-13-78. [Google Scholar] [CrossRef]
Li W, Li G, She W, Hu X, Wu X. (2019). Targeted antitumor activity of Ginsenoside (Rg1) in paclitaxel-resistant human nasopharyngeal cancer cells are mediated through activation of autophagic cell death, cell apoptosis, endogenous ROS production, S phase cell cycle arrest and inhibition of m-TOR/PI3K/AKT signalling pathway. Journal of BUON 24: 2056–2061. [Google Scholar]
Ma YM, Ibeanu G, Wang LY, Zhang JZ, Chang Y, Dong JD, Li PA, Jing L. (2017). Selenium suppresses glutamate-induced cell death and prevents mitochondrial morphological dynamic alterations in hippocampal HT22 neuronal cells. BMC Neuroscience 18: 15. DOI 10.1186/s12868-017-0337-4. [Google Scholar] [CrossRef]
Madrigal-Matute J, Cuervo AM. (2016). Regulation of liver metabolism by autophagy. Gastroenterology 150: 328–339. DOI 10.1053/j.gastro.2015.09.042. [Google Scholar] [CrossRef]
Medici V, Huster D. (2017). Animal models of Wilson disease. Handbook of Clinical Neurology 142: 57–70. [Google Scholar]
Meng H, Wang L, He J, Wang Z. (2016). The protective effect of gangliosides on lead (Pb)-induced neurotoxicity is mediated by autophagic pathways. International Journal of Environmental Research and Public Health 13: 365. DOI 10.3390/ijerph13040365. [Google Scholar] [CrossRef]
Parzych KR, Klionsky DJ. (2014). An overview of autophagy: Morphology, mechanism, and regulation. Antioxidants & Redox Signaling 20: 460–473. DOI 10.1089/ars.2013.5371. [Google Scholar] [CrossRef]
Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W, Zheng H, Bosenberg MW, Mehnert JM, Guo JY, Lattime E, Rabinowitz JD, White E. (2018). Autophagy maintains tumour growth through circulating arginine. Nature 563: 569–573. DOI 10.1038/s41586-018-0697-7. [Google Scholar] [CrossRef]
Polishchuk EV, Merolla A, Lichtmannegger J, Romano A, Indrieri A, Ilyechova EY, Concilli M, Cegli RD, Crispino R, Mariniello M, Petruzzelli R, Ranucci G, Iorio R, Pietrocola F, Einer C, Borchard S, Zibert A, Schmidt HH, Schiavi ED, Puchkova LV, Franco B, Kroemer G, Zischka H, Polishchuk RS. (2019). Activation of autophagy, observed in liver tissues from patients with Wilson disease and from ATP7B-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology 156: 1173–1189. DOI 10.1053/j.gastro.2018.11.032. [Google Scholar] [CrossRef]
Przybylkowski A, Gromadzka G, Wawer A, Grygorowicz T, Cybulska A, Czlonkowska A. (2013). Intestinal expression of metal transporters in Wilson’s disease. BioMetals 26: 925–934. DOI 10.1007/s10534-013-9668-5. [Google Scholar] [CrossRef]
Sharma Y, Liu J, Kristian KE, Follenzi A, Gupta S. (2018). In Atp7b−/− mice modeling Wilson’s disease liver repopulation with bone marrow-derived myofibroblasts or inflammatory cells and not hepatocytes is deleterious. Gene Expression The Journal of Liver Research 19: 15–24. DOI 10.3727/105221618X15320123457380. [Google Scholar] [CrossRef]
Shi Z, Liang XL, Lu BX, Pan SY, Chen X, Tang QQ, Wang Y, Huang F. (2005). Diminution of toxic copper accumulation in toxic milk mice modeling Wilson disease by embryonic hepatocyte intrasplenic transplantation. World Journal of Gastroenterology 11: 3691–3695. DOI 10.3748/wjg.v11.i24.3691. [Google Scholar] [CrossRef]
Ueno T, Komatsu M. (2017). Autophagy in the liver: Functions in health and disease. Nature Reviews Gastroenterology & Hepatology 14: 170–184. DOI 10.1038/nrgastro.2016.185. [Google Scholar] [CrossRef]
Weiskirchen S, Kim P, Weiskirchen R. (2019). Determination of copper poisoning in Wilson’s disease using laser ablation inductively coupled plasma mass spectrometry. Annals of Translational Medicine 7: S72. DOI 10.21037/atm.2018.10.67. [Google Scholar] [CrossRef]
Zheng Y, Hou J, Liu J, Yao M, Li L, Zhang B, Zhu H, Wang Z. (2014). Inhibition of autophagy contributes to melatonin-mediated neuroprotection against transient focal cerebral ischemia in rats. Journal of Pharmacological Sciences 124: 354–364. DOI 10.1254/jphs.13220FP. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |