| Computer Modeling in Engineering & Sciences |
DOI: 10.32604/cmes.2021.016622
ARTICLE
Predicting Genotype Information Related to COVID-19 for Molecular Mechanism Based on Computational Methods
1Jiangsu Key Laboratory of Big Data Security & Intelligent Processing School of Computer Science, Nanjing University of Posts and Telecommunications, Nanjing, 210046, China
2Smart Health Big Data Analysis and Location Services Engineering Laboratory of Jiangsu Province, Nanjing, 210046, China
3College of Computer Science and Technology, Nanjing Forestry University, Nanjing, 210037, China
4Zhejiang Engineering Research Center of Intelligent Medicine, Wenzhou, 325035, China
*Corresponding Author: Lejun Gong. Email: glj98226@163.com
Received: 12 March 2021; Accepted: 10 June 2021
Abstract: Novel coronavirus disease 2019 (COVID-19) is an ongoing health emergency. Several studies are related to COVID-19. However, its molecular mechanism remains unclear. The rapid publication of COVID-19 provides a new way to elucidate its mechanism through computational methods. This paper proposes a prediction method for mining genotype information related to COVID-19 from the perspective of molecular mechanisms based on machine learning. The method obtains seed genes based on prior knowledge. Candidate genes are mined from biomedical literature. The candidate genes are scored by machine learning based on the similarities measured between the seed and candidate genes. Furthermore, the results of the scores are used to perform functional enrichment analyses, including KEGG, interaction network, and Gene Ontology, for exploring the molecular mechanism of COVID-19. Experimental results show that the method is promising for mining genotype information to explore the molecular mechanism related to COVID-19.
Keywords: COVID-19; SARS-CoV-2; computational method; bioinformatics; genotype; machine learning
Novel coronavirus disease 2019 (COVID-19), caused by a novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is currently ravaging the world. It is the seventh known coronavirus that can infect humans. COVID-19 is highly infectious and can cause serious complications, posing a great threat to global public safety [1]. Publicly available data indicate a fatality rate of 3% [2]. One country after another has taken steps to tackle the disease. The EU has mobilized a research fund of EUR 10 million to effectively manage patients and public health preparedness and response [3]. The UK Government has invested £20 million to help develop a vaccine against COVID-19 [4]. According to Bloomberg news, U.S. President Donald Trump signed a $7.8 billion emergency spending bill for COVID-19 epidemic [5]. The source of the virus is unknown, and a specific medicine for the disease it causes remains unavailable. Many computational methods are also used in the research of COVID-19. Kumari et al. [6] developed forecasting models and predicted the number of confirmed, recovered, and death cases in India caused by COVID-19 by using statistical models with correlation coefficients and multiple linear regression. The replication, expression, and regulation of the virus depend on the host system, which reflects its particularity in genetic composition, gene expression mode, and interaction with other organisms. Therefore, researchers are trying to understand the genetic mechanism of COVID-19. The downregulation of ACE2, which is the SARS-CoV-2 receptor, is an important aspect of SARS-CoV-2 mortality suffered by elderly men [7]. This study highlighted that the S1 domain of COVID-19 spike glycoprotein potentially interacts with the human CD26, which is a key immunoregulatory factor for hijacking and virulence [8]. Tai et al. identified the receptor-binding domain (RBD) in SAR-Cov-2 S protein and found that the RBD protein binds strongly to human ACE2 receptors. Thus, the RBD protein could be developed as a viral attachment inhibitor and vaccine [9]. This work reveals that coronavirus engages papain-like proteases (PLPs) to escape from the innate antiviral response of the host by inhibiting p53-IRF7-IFNβ signaling [10].
With the rapid development of information technology and the continuous generation of high-throughput biological data, an endless stream of coronavirus-related research results, published experimental data, and biomedical literature has opened a door to the computational study of this viral disease [11–13]. Despite the many applications of biomedical text mining [14–17], few studies focused on COVID-19 related to text mining, and most of these studies are clinical and experimental works [18–20].
This paper proposes a prediction method for mining genotype information related to COVID-19 by using computational methods based on biomedical text mining. The method obtains seed genes related to COVID-19 and candidate genes by literature. Furthermore, it uses bioinformatics technique and statistical method for mining genotype information about the molecular mechanism of COVID-19. The novelty of this paper is as follows:
(1) Proposing an effective method for mining gene information related to COVID-19;
(2) Predicting gene information related to COVID-19 from text mining based on computation;
(3) Analyzing the molecular mechanism of COVID-19 on the basis of genotype information.
Details are shown in the following sections.
Seed and candidate genes were obtained from biomedical databases and literature, respectively. Then, the similarity matrix between the candidate and seed genes was calculated on the basis of the semantic similarity of genetic terms. The matrix was taken as the original data set for training in the random forest regression model. The genes with high scores were prioritized as the final candidate genes to analyze genotype information. Finally, the molecular mechanism of pathogenic genes was explored to understand the etiology and thus elucidate the molecular mechanism of COVID-19. The pipeline is described in Fig. 1.
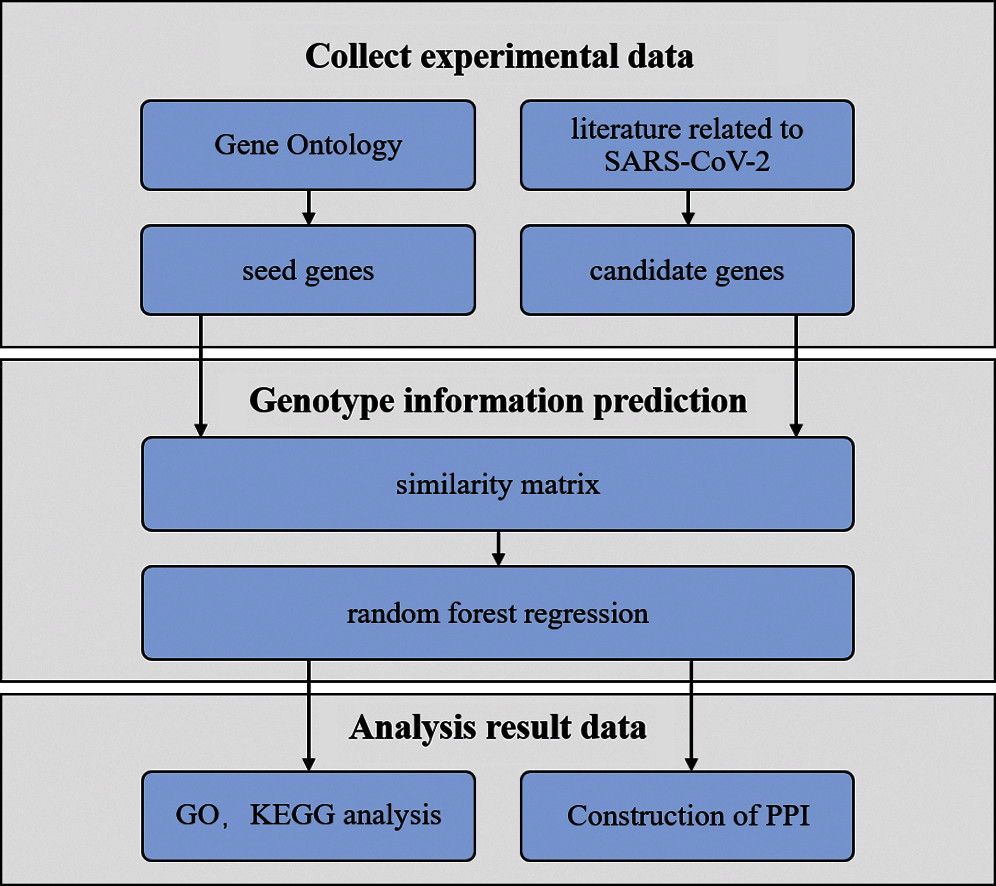
Figure 1: Pipeline of this work
As shown in Fig. 1, experimental data were collected from Gene Ontology (GO) and literature. The similarity matrix was taken as the original data set for training in the random forest regression model to predict genotype information. Functional enrichment analysis by KEGG, interacts network, and GO was performed to explore the molecular mechanism related to SARS-CoV-2.
GO [21] (http://geneontology.org/covid-19.html) is a knowledge base that describes the function of genes based on evidence in the scientific literature. To assist global research on the SARS-CoV-2 virus, the GO Consortium is integrating SARS-CoV-2 genes for the curation and reuse of recent articles on SARS-CoV-2. In this study, the human genes that may be related to SARS-CoV-2 infection published in the GO database were taken as seed genes, including 327 genes, on October 21, 2020.
The biomedical literature related to SARS-CoV-2 was extracted from the PubMed database with E-Utilities (http://eutils.ncbi.nlm.nih.gov/corehtml/query/static/eutils_help.html) by using the key word “SARS-CoV-2” on November, 10 2020. The genes mined from this literature were taken as initial candidate genes, including 315 genes. The human genome data set was derived from HUGO Gene Nomenclature Committee [22] (HGNC; https://www.genenames.org/).
2.2 Calculation of Semantic Similarity between Genes
Genes have abundant functional information. Predicting gene function by analyzing the functional information of genes has become a new research direction. In general, if two gene products have similar functions, then they have similar GO annotation terms. Therefore, the related functions of unknown genes can be predicted by analyzing gene terminology. Many methods have been proposed for calculating the semantic similarity between genes [23,24]. Most of these methods depend on the results of calculating the semantic similarity between genetic terms. Typical methods for calculating the similarity between genetic terms, such as Resnik [25], are based on the idea that the semantic similarity of two terms is related to the corresponding lowest common ancestor node, and the similarity is calculated by the ancestor node with the most information in the lowest common ancestor set. The formula is shown in Eq. (1):
where
where
A typical method for calculating the semantic similarity among genes is the optimal matching algorithm proposed by Couto et al. [27]. This method defines the semantic similarity between genes as the maximum semantic similarity between terms in the annotations corresponding to genes. Instead of computing the maximum pairwise GO term similarity, one may also take the average here. Given two genes g and g’ annotated with GO terms t1,…tn and t’1,…, t’m, the similarity between g and g’ is denoted as Eq. (3):
Random forest algorithm (RFA) is a classification and prediction model proposed by Leo Breiman [28]. As a multi-classifier algorithm based on ensemble learning, RFA is characterized by fast running speed, fewer parameters to be adjusted, and efficient processing of large sample data. Based on the construction of bagging integration with decision trees that are used as learners, this algorithm introduces randomization. The following describes the decision trees, bagging, and randomization.
Decision tree is a tree-shaped classifier. Leaf nodes correspond to decision results; each internal node represents a feature-based test; the sample set of each node is divided into sub-nodes according to the result of feature test. The root node contains a full set of samples. The construction of the decision tree depends on the training sample data and the characteristics used to divide each internal node.
Classification and regression tree uses the Gini Index to select the partitioning attribute. The purity of dataset D can be measured by the Gini value. The smaller Gini(D) is, the higher the purity is. The formula is shown in Eq. (4):
The Gini index of attribute a is calculated using Eq. (5):
In the candidate attribute set A, the attribute with the minimum Gini index after partition is selected as the optimal partition attribute, which means
Assuming that the number of samples in the original training set is N, a new training set can be formed by randomly extracting N samples from the original training set by using Bagging method. It can be calculated that about 37% of the data generated in each new training set may not be elected, and this part of the data is called out-of-bag (OOB). OOB can be used as test data to estimate the generalization performance of the decision tree, that is, out-of-bag estimation.
The traditional decision tree selects the best feature from the feature set of the current node when choosing the split feature, while in RFA, features are randomly selected for node splitting. There are two ways: the first is to determine the number of candidate features F for each splitting, and then randomly select F features from the full feature set, and then split the nodes according to the optimal splitting criterion; the second method is to randomly select L features, then select the coefficients randomly to make a linear combination of them to generate F new features, and then split the nodes according to the optimal splitting criterion.
The principle of random forest is summarized as follows: In the first step, samples were extracted with bagging method to form several training sets. The second step is to randomly select the characteristics of each tree during the growth process to split the internal nodes. Then, the steps are repeated to maximize the growth of each tree. Finally, the randomly growing trees constitute the forest, and the new data are predicted based on the generated random forest [29].
Genes related to COVID-19 were optimized by twice prioritizations. The initial candidate genes were obtained from biomedical literature related to the key word “SARS-CoV-2.” The seed and initial candidates were constructed as the semantic similar matrix that serves as the input of RFA for the second prioritizations. The optimized genes that exceed the threshold were further analyzed by bioinformatics analysis. Functional enrichment analysis was performed for the predicted pathogenic genes and construction of PPI and hub gene identification.
3.1 Matrix of Semantic Similarity
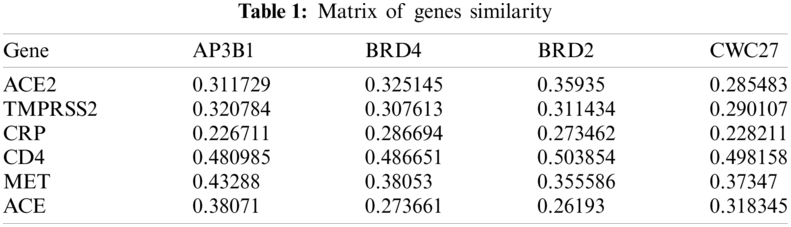
The GOSim packet (version 1.28.0; https://www.bioconductor.org/packages/release/bioc/html/GOSim.html) [30] in R was used to calculate the similarity between seed genes and some genes related to COVID-19 and obtain a matrix of 69 * 327; to calculate the similarity between seed genes and randomly selected human genes and obtain a matrix of 80 * 327; and to calculate the similarity between seed genes and candidate genes and obtain a matrix of 315 * 327. Some of the results are shown in Tab. 1.
The similarity matrix was obtained in the previous step, which was used as the original data set and was preprocessed. Then, the similarity between a gene and each seed gene was taken as the influencing factor of whether or not it was a disease-related gene. That is, each sample had 327 independent variables. The dependent variable Y value of the known disease-related gene sample set was set to 1. After removing the seed and candidate genes, human genes were randomly selected as a disease-independent sample set with a Y threshold of 0.9. The Synthetic Minority Over-sampling Technique [31] algorithm was used to up-sample the data.
The Mean Square Error (MSE), Mean Absolute Error (MAE), and coefficient of determination (R2) were used to measure the fitting effect of the model. The results are with an MSE of 0.0415, a MAE of 0.1418, and an R2 of 0.8291 aiming at the size of training set with 97 genes and testing set with 52 genes.
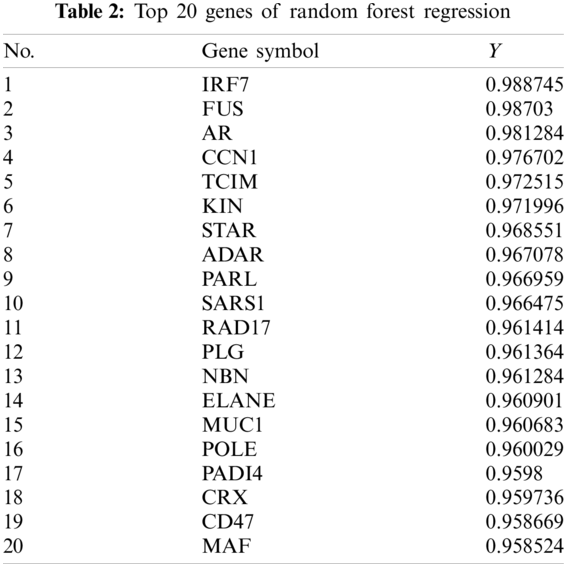
After inputting the data into the model, we obtained the predicted results, some of which are shown in Tab. 2. A total of 125 genes with a Y value > 0.9 were selected as the disease-related genes.
WEB-based GEne SeT AnaLysis Toolkit (WebGestalt; http://www.webgestalt.org/) [32] is a popular tool for the interpretation of gene lists derived from large-scale omics studies. GO terminology, which includes biological process, molecular function (MF), and cellular component (CC), and Kyoto Encyclopedia of Genes and Genomes (KEGG) [33] pathway enrichment analyses were performed to predict pathogenic genes using WebGestalt.
To understand the GO categories of predicted disease genes, we placed the seed and predicted genes into WebGestalt to perform GO analysis. GO analysis showed that the seed and predicted genes mainly participate in metabolic process, biological regulation, and response to stimulus. Most of the gene sets are located in membrane-enclosed lumen and nucleus. Figs. 2–4 display the results of BP, CC, and MF enrichment, respectively.
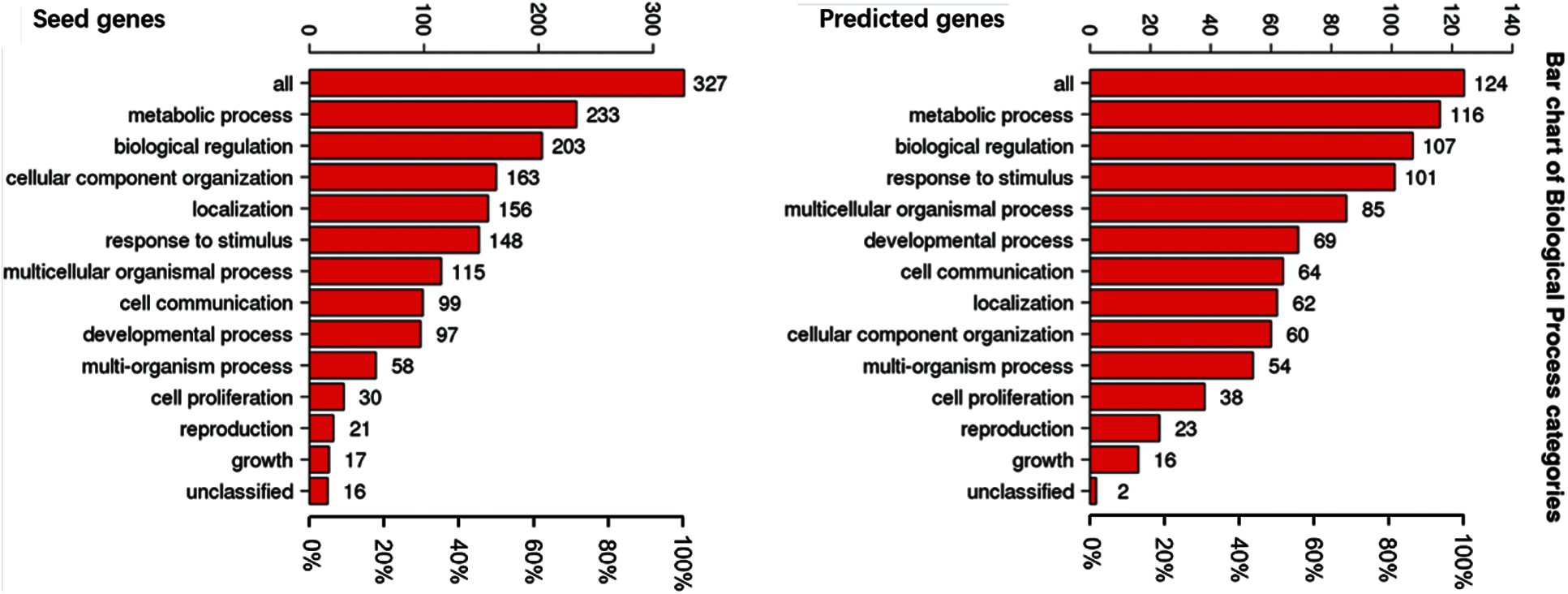
Figure 2: Biological process enrichment
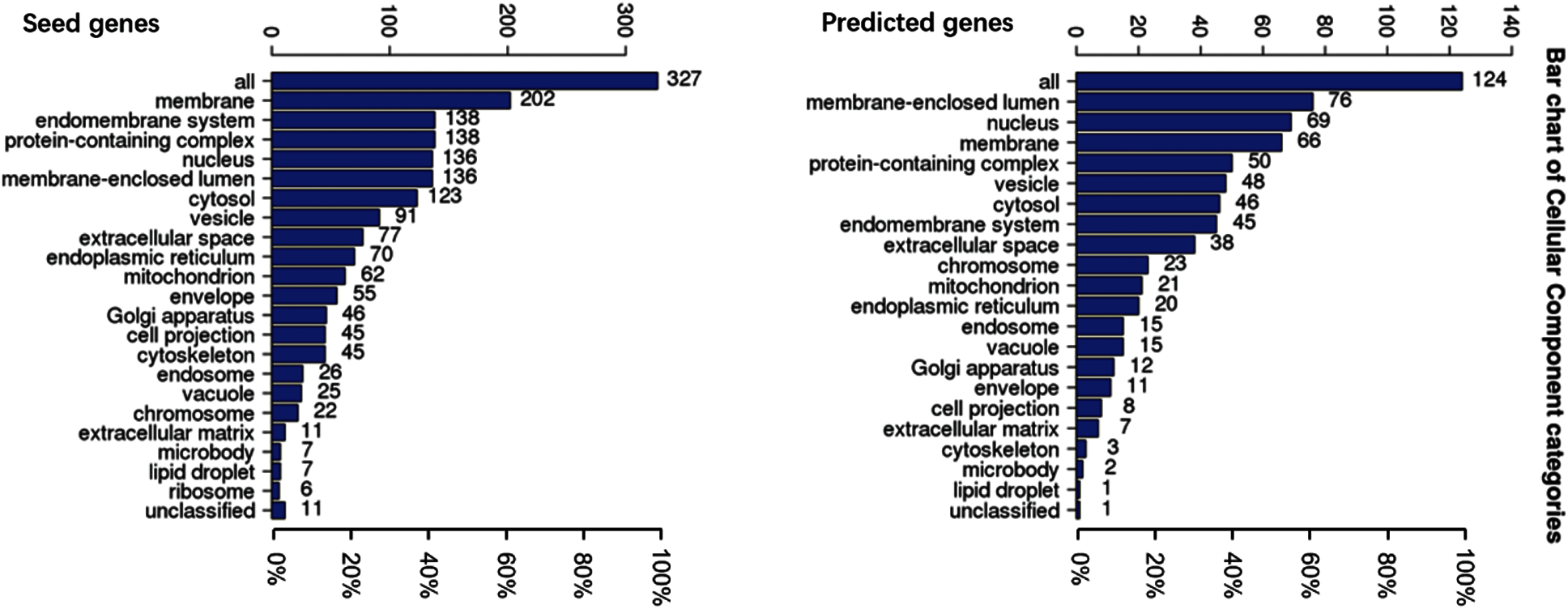
Figure 3: Cellular component enrichment
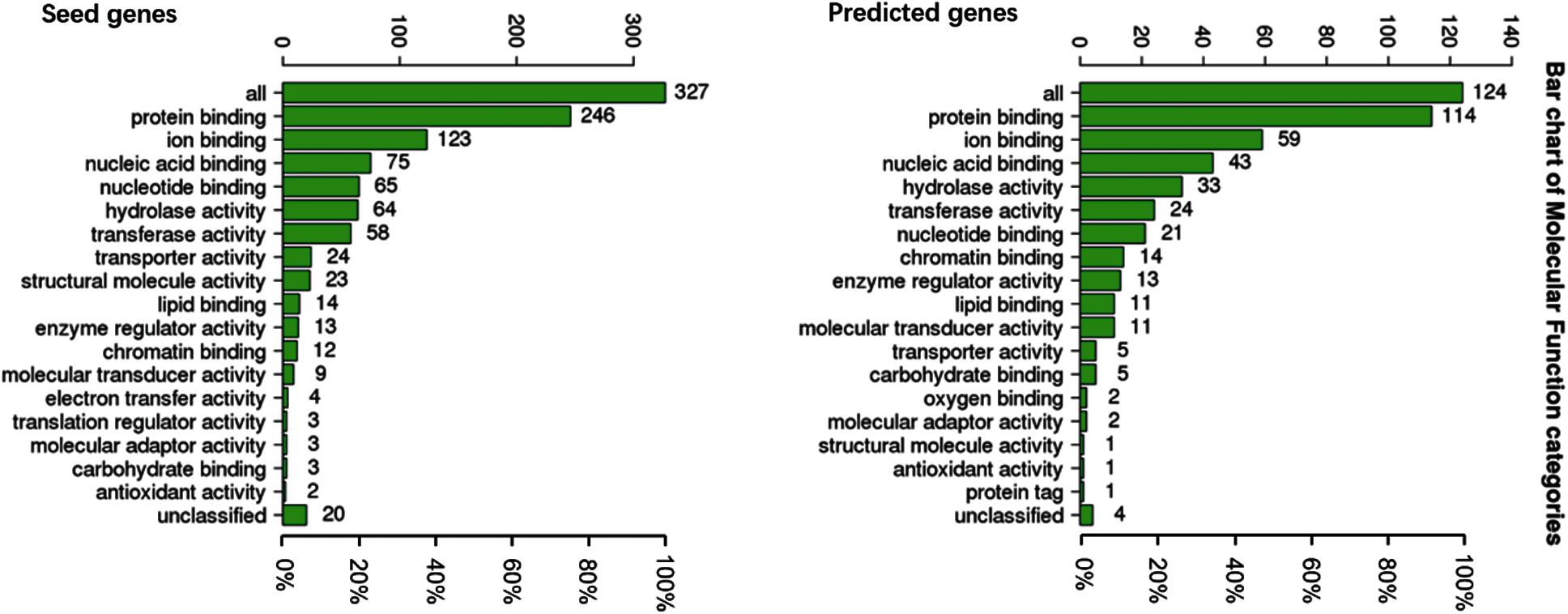
Figure 4: Molecular function enrichment
KEGG analysis indicated that the predicted genes are associated with Influenza A, virus infection, and RIG-I-like receptor signaling pathway. The results are shown in Fig. 3. GO and KEGG analyses of these genes provide a reference for understanding the molecular mechanism of the unknown samples, which could find new genes related to COVID-19.
From Figs. 2–4, GO analysis shows that the seed and predicted genes are mainly enriched in metabolic process, biological regulation, membrane-enclosed lumen and nucleus, and protein binding, which indicate that they have similar molecular mechanisms between the seed and predicted genes.
The seed and predicted genes were subjected to KEGG analysis, and Fig. 5 shows the result of KEGG analysis.
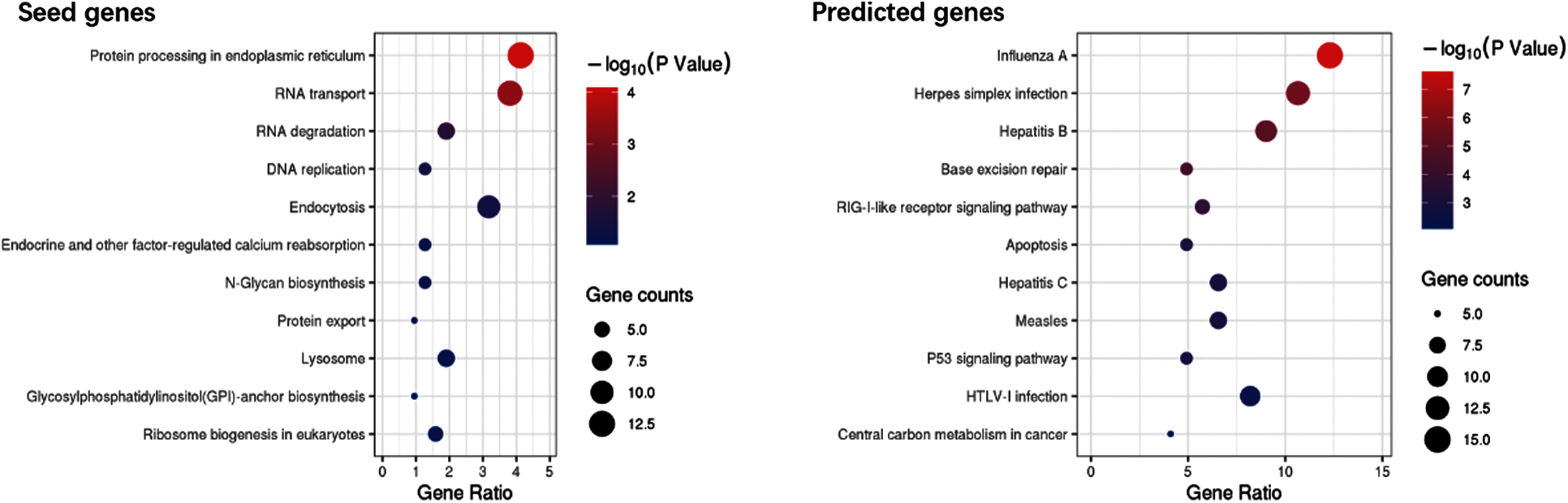
Figure 5: Bubble plot of KEGG pathway enrichment
The results of the KEGG pathway enrichment in Fig. 5 indicated that the predicted genes are associated with Influenza A, virus infection, and RIG-I-like receptor signaling pathway.
GO analysis revealed that the seed and predicted genes have similar molecular mechanisms. KEGG analysis indicated that the predicted genes are involved with the pathways of influenza A and virus infection. GO and KEGG analyses of these genes provide a reference for understanding the molecular mechanism of the unknown samples, which could find new genes related to COVID-19.
We used the Search Tool for the Retrieval of Interacting Genes (STRING) database [34], which is an online tool for exploring and analyzing information about protein interactions, was used to explore the relationship between disease-related genes. STRING was used to map the prediction and seed genes to evaluate the interaction. Then, the PPI network was constructed and visualized using Cytoscape [35] software (version 3.5.1; https://www.cytoscape.org). The hub genes in the network were identified by the plug-in Molecular Complex Detection (MCODE; version 1.5.1) [36] with a degree cutoff = 6, haircut = on, node score cut-off = 0.2, k-core = 2, and max. depth = 100 to determine key elements.
PPI networks were constructed using STRING and Cytoscape to study the connections between the identified and seed genes, and predict the associations of protein functions related to the identified genes. The network that is composed of 125 predicted disease genes and 327 seed genes is shown in Fig. 6 containing 447 nodes and 2016 edges with an average node degree of 9.02, a local clustering coefficient of 0.384, and a significant enrichment of PPI (P < 0.001).
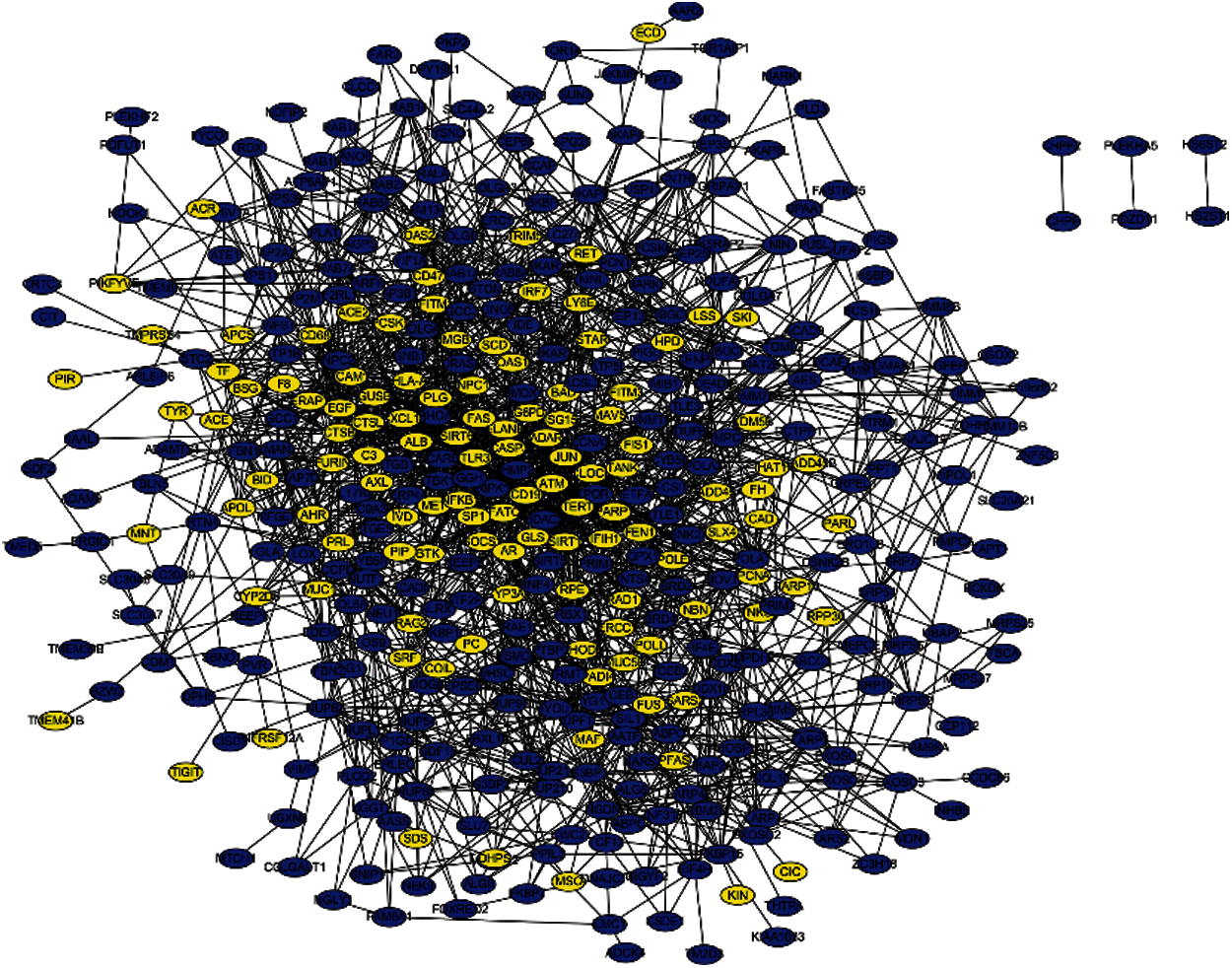
Figure 6: PPI network containing seed and predicted genes
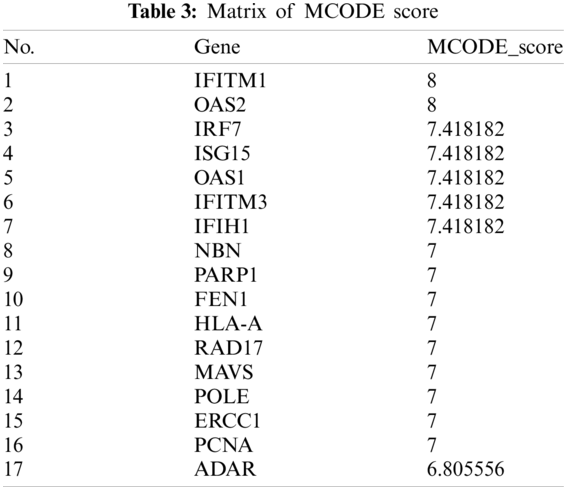
The PPI network that is composed of 125 predicted disease genes contains 124 nodes and 514 edges with an average node degree of 8.29, a local clustering coefficient of 0.521, and a significant enrichment of PPI (P < 0.001), as shown in Fig. 7. The top 17 hub genes with the highest connective degree, RAD17, POLE, PCNA, PARP1, OAS2, OAS1, NBN, MAVS, ISG15, IRF7, IFITM3, IFITM1, IFIH1, HLA-A, FEN1, RCC1, and ADAR, were selected by MCODE from Fig. 7. These hub genes were also primarily associated with DNA replication, viral genome replication, and Type I interferon signaling pathway. The most significant module identified in the PPI network is shown in Fig. 8.
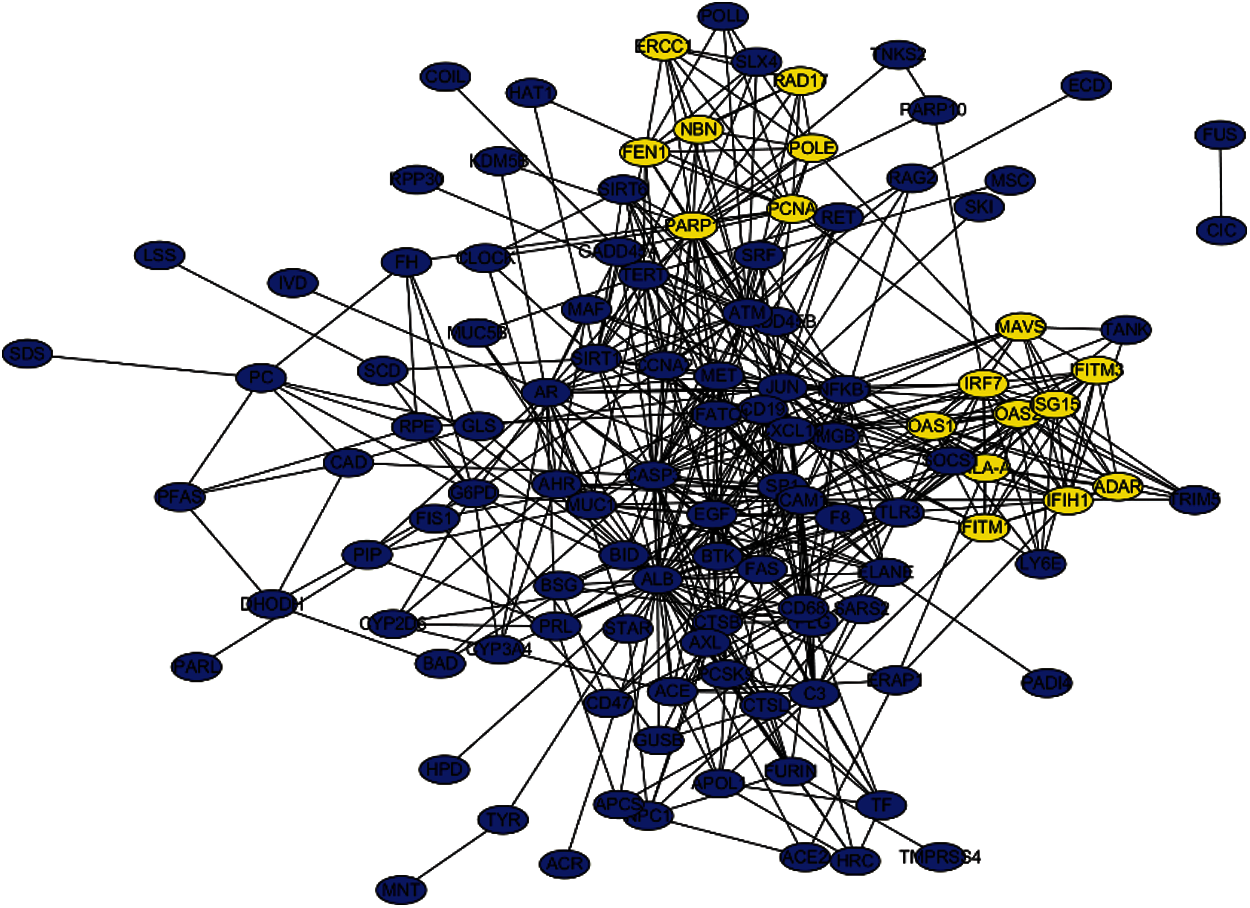
Figure 7: PPI network of the predicted genes
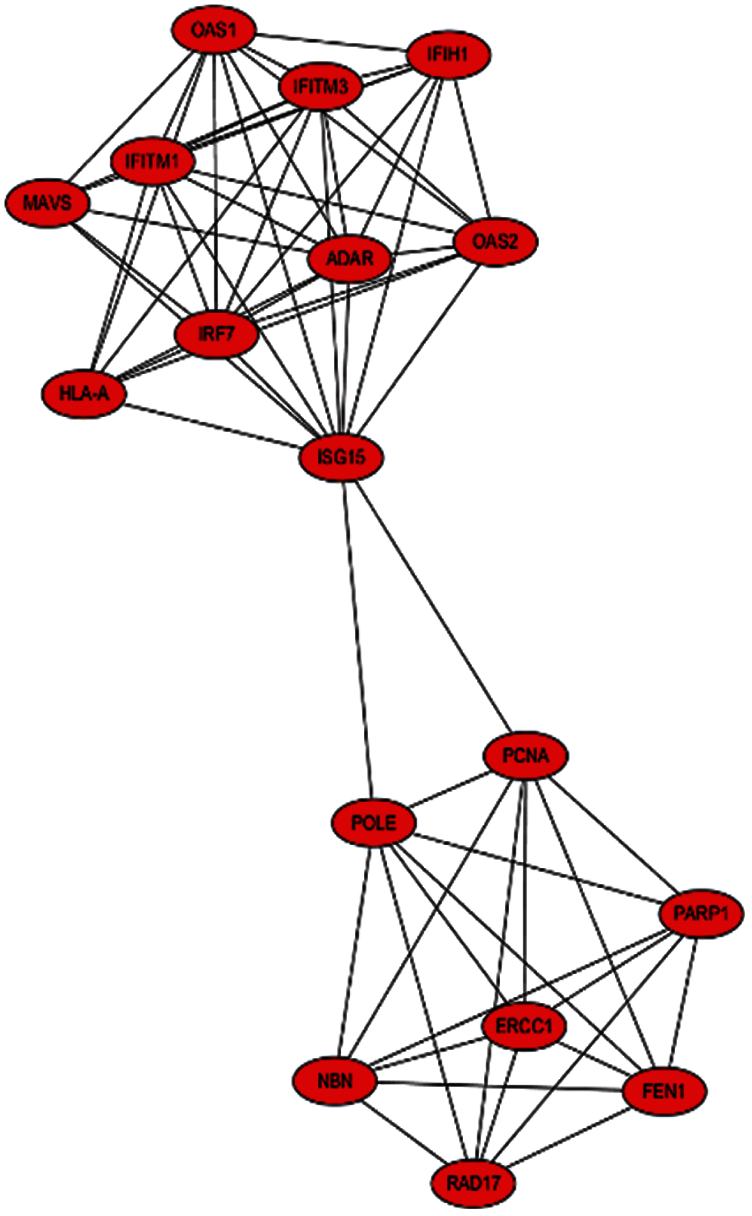
Figure 8: Most significant module from Fig. 7
The MCODE scores of the top 17 hub genes are shown in Tab. 3. They are in the most significant module identified in the PPI network related to the predicted genes.
The 17 genes were further analyzed by literature evidence to determine the validity of our method. Studies have shown that SARS-CoV-2 infection is characterized by a high mortality rate from age-related diseases in older men [7,37]. Two host receptors for COVID-19, CD26 [8] and ACE-2 (angiotensin-converting enzyme 2) [9], are associated with aging. Krishna et al. [38] found that transcriptional changes of target genes regulating mitochondrial function (such as FEN1), cell senescence (such as PCNA), and telomere loss (such as RAD17, NBN, and PARP1) in the pathobiological process of COPD and IPF were related to changes in the ACE2-TMPRSS2-Furin-DPP4 axis of COVID-19. Aging plays an important role in SARS-CoV-2 infection. Therefore, anti-aging drugs possibly have a positive effect on the treatment and prevention of COVID-19. The interferon-stimulated gene (ISG) family includes IFITM3, IFITM1, IFIH1, OAS2, OAS3, IRF7, MAVS, and so on. IFN-I (I type of interferon) response plays a key role in antiviral infection. It can prevent the virus by inducing the expression of ISGs [39]. SARS-CoV-2 induces a strong interferon response [40]. Kristel et al. [41] found that ISGs are significantly upregulated in bronchoalveolar lavage fluid from patients with COVID-19. SARS-CoV can use its structural and non-structural proteins to counter the effect of IFN and suppress innate immunity [42]. For example, SARS-CoV ORF-9b manipulates host cell mitochondria and mitochondrial function and inhibits MAVS signaling to suppress innate immunity [43]. Coronavirus PLPs have been identified as suppressors of the innate immune response. The ISG15-dependent activation of MDA5 is antagonized through direct de-ISGylation mediated by the PLPs of SARS-CoV-2. IRF7, as a target gene of p53, mediates the p53-directed production of Type I interferon. By promoting p53 degradation, PLPs inhibit the p53-mediated antiviral response to help evade host innate immunity [44].
According to the above analysis results, the top 17 hub genes are more or less related to SARS-CoV-2.
COVID-19 is a serious threat to people's health and life safety currently. The acquisition of its genetic information is important. The novelty of this work is (1) proposing an effective method for mining genotype information related to COVID-19; (2) predicting gene information related to COVID-19 from text mining based on computation; and (3) analyzing molecular mechanism of genotype information of COVID-19. Initially, 327 seed genes were obtained from the gene ontology database based on evidence. The initial 315 candidates were obtained from biomedical literature with the keyword “SARS-CoV-2” by text mining. Then, the semantic similarity matrix between seeds and candidates was constructed. The results were processed using RFA. The 125 disease genes with the Y threshold of 0.9 were prioritized. GO and pathway analyses based on the WebGestalt tool were performed to analyze further the biological functions of these disease genes. Enrichment analysis results indicated that these genes were mainly enriched in DNA replication, type I interferon signaling pathway, and DNA repair. KEGG pathway analysis indicated that these genes were mainly enriched in Influenza A, virus infection, and RIG-I-like receptor signaling pathway. These results contribute to further understanding the possible roles of these genes in the occurrence and development of COVID-19. Basing on these 125 predicted disease genes, we constructed a PPI network. We identified 17 hub genes in this network, which were believed to be closely related to the pathogenesis of COVID-19 based on literary evidence. The 125 predicted disease genes that may be correlated with COVID-19 could be analyzed using the computational approach. This method may provide a new clue for investigating the potential biomarkers and biological genetic mechanisms of COVID-19. The results also show it is promising for mining genotype information for exploring its molecular mechanism for further developing the potential diagnosis and therapeutic intervention methods of COVID-19.
Further works would be to detect advanced technologies to extract more accurate genotype information related to COVID-19. In addition, the phenotypic information related to COVID-19 will be mined so that genotypic and phenotypic information can be combined to elucidate the molecular mechanism and pathogenesis of COVID-19.
Funding Statement: This research is supported by the National Natural Science Foundation of China (Grant Nos. 61502243, 61802193), Natural Science Foundation of Jiangsu Province (BK20170934), Zhejiang Engineering Research Center of Intelligent Medicine under 2016E10011, China Postdoctoral Science Foundation (2018M632349), NUPTSF (NY217136), Foundation of Smart Health Big Data Analysis and Location Services Engineering Laboratory of Jiangsu Province (SHEL221-001), and Natural Science Foundation of the Higher Education Institutions of Jiangsu Province in China (16KJD520003).
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
1. Ahn, D. G., Shin, H. J., Kim, M. H., Lee, S., Kim, H. S. et al. (2020). Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19). Journal of Microbiolog and Biotechnology, 30(3), 313–324. DOI 10.4014/jmb.2003.03011. [Google Scholar] [CrossRef]
2. Sohrabi, C., Alsafi, Z., O'Neill, N., Khan, M., Kerwan, A. et al. (2020). World health organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). International Journal of Surgery, 76, 71–76. DOI 10.1016/j.ijsu.2020.02.034. [Google Scholar] [CrossRef]
3. Goniewicz, K., Khorram-Manesh, A., Hertelendy, A. J., Goniewicz, M., Naylor, K. et al. (2020). Current response and management decisions of the european union to the COVID-19 outbreak: A review. Sustainability, 12(9), 3838. DOI 10.3390/su12093838. [Google Scholar] [CrossRef]
4. Ledford, H., Cyranoski, D., van Noorden, R. (2020). The UK has approved a COVID vaccine—Here's what scientists now want to know. Nature, 588(7837), 205–206. DOI 10.1038/d41586-020-03441-8. [Google Scholar] [CrossRef]
5. Lancet, T. (2020). Global governance for COVID-19 vaccines. Lancet (London, England), 395(10241), 1883. DOI 10.1016/S0140-6736(20)31405-7. [Google Scholar] [CrossRef]
6. Kumari, R., Kumar, S., Poonia, R. C., Singh, V., Raja, L. et al. (2021). Analysis and predictions of spread, recovery, and death caused by COVID-19 in India. Big Data Mining and Analytics, 4(2), 65–75. DOI 10.26599/BDMA.2020.9020013. [Google Scholar] [CrossRef]
7. Bonafè, M., Prattichizzo, F., Giuliani, A., Storci, G., Sabbatinelli, J. et al. (2020). Inflamm-aging: Why older men are the most susceptible to SARS-CoV-2 complicated outcomes. Cytokine & Growth Factor Reviews, 53, 33–37. DOI 10.1016/j.cytogfr.2020.04.005. [Google Scholar] [CrossRef]
8. Vankadari, N., Wilce, J. A. (2020). Emerging Wuhan (COVID-19) coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerging Microbes & Infections, 9(1), 601–604. DOI 10.1080/22221751.2020.1739565. [Google Scholar] [CrossRef]
9. Tai, W., He, L., Zhang, X., Pu, J., Voronin, D. et al. (2020). Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cellular & Molecular Immunology, 17(6), 613–620. DOI 10.1038/s41423-020-0400-4. [Google Scholar] [CrossRef]
10. Yuan, L., Chen, Z., Song, S., Wang, S., Tian, C. et al. (2015). P53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. Journal of Biological Chemistry, 290(5), 3172–3182. DOI 10.1074/jbc.M114.619890. [Google Scholar] [CrossRef]
11. Wang, L. L., Lo, K. (2020). Text mining approaches for dealing with the rapidly expanding literature on COVID-19. Briefings in Bioinformatics, 22(2), 781–799. DOI 10.1093/bib/bbaa296. [Google Scholar] [CrossRef]
12. Umar, M., Sabir, Z., Raja, M. A. Z., Shoaib, M., Gupta, M. et al. (2020). A stochastic intelligent computing with neuro-evolution heuristics for nonlinear SITR system of novel COVID-19 dynamics. Symmetry, 12(10), 1628. DOI 10.3390/sym12101628. [Google Scholar] [CrossRef]
13. Umar, M., Sabir, Z., Raja, M. A. Z., Amin, F., Saeed, T. et al. (2021). Integrated neuro-swarm heuristic with interior- point for nonlinear SITR model for dynamics of novel COVID-19. Alexandria Engineering Journal, 60(3), 2811–2824. DOI 10.1016/j.aej.2021.01.043. [Google Scholar] [CrossRef]
14. Rodriguez-Esteban, R., Bundschus, M. (2016). Text mining patents for biomedical knowledge. Drug Discovery Today, 21(6), 997–1002. DOI 10.1016/j.drudis.2016.05.002. [Google Scholar] [CrossRef]
15. Gong, L., Yang, R., Yan, Q., Sun, X. (2013). Prioritization of disease susceptibility genes using LSM/SVD. IEEE Transactions on Biomedical Engineering, 60(12), 3410–3417. DOI 10.1109/TBME.2013.2257767. [Google Scholar] [CrossRef]
16. Gong, L., Yan, Y., Xie, J., Liu, H., Sun, X. (2012). Prediction of autism susceptibility genes based on association rules. Journal of Neuroscience Research, 90(6), 1119–1125. DOI 10.1002/jnr.23015. [Google Scholar] [CrossRef]
17. Gong, L., Zhang, Z., Chen, S. (2020). Clinical named entity recognition from Chinese electronic medical records based on deep learning pretraining. Journal of Healthcare Engineering, 2020, 8829219. DOI 10.1155/2020/8829219. [Google Scholar] [CrossRef]
18. Chen, Y., Klein, S. L., Garibaldi, B. T., Li, H., Wu, C. et al. (2021). Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Research Reviews, 65, 101205. DOI 10.1016/j.arr.2020.101205. [Google Scholar] [CrossRef]
19. Yüce, M., Filiztekin, E., Özkaya, K. G. (2021). COVID-19 diagnosis---A review of current methods. Biosensors and Bioelectronics, 172, 112752. DOI 10.1016/j.bios.2020.112752. [Google Scholar] [CrossRef]
20. Kang, H., Wang, Y., Tong, Z., Liu, X. (2020). Retest positive for SARS-CoV-2 RNA of “recovered” patients with COVID-19: Persistence, sampling issues, or re-infection? Journal of Medical Virology, 92(11), 2263–2265. DOI 10.1002/jmv.26114. [Google Scholar] [CrossRef]
21. Gene Ontology Consortium (2015). Gene ontology consortium: Going forward. Nucleic Acids Research, 43(D1), D1049–D1056. DOI 10.1093/nar/gku1179. [Google Scholar] [CrossRef]
22. Eyre, T. A., Ducluzeau, F., Sneddon, T. P., Povey, S., Bruford, E. A. et al. (2006). The HUGO gene nomenclature database. Nucleic Acids Research, 34(Database Issue), 319–321. [Google Scholar]
23. Schlicker, A., Domingues, F. S., Rahnenführer, J., Lengauer, T. (2006). A new measure for functional similarity of gene products based on gene ontology. BMC Bioinformatics, 7(1), 1–16. DOI 10.1186/1471-2105-7-1. [Google Scholar] [CrossRef]
24. Pesquita, C., Faria, D., Bastos, H., Ferreira, A. E., Falcão, A. O. et al. (2008). Metrics for GO based protein semantic similarity: A systematic evaluation. BMC Bioinformatics, 9(5), 1–16. DOI 10.1186/1471-2105-9-S5-S4. [Google Scholar] [CrossRef]
25. Resnik, P. (1999). Semantic similarity in a taxonomy: An information-based measure and its application to problems of ambiguity in natural language. Journal of Artificial Intelligence Research, 11, 95–130. DOI 10.1613/jair.514. [Google Scholar] [CrossRef]
26. Lord, P. W., Stevens, R. D., Brass, A., Goble, C. A. (2002). Semantic similarity measures as tools for exploring the gene ontology. Biocomputing, 2003, 601–612. DOI 10.1142/5149. [Google Scholar] [CrossRef]
27. Couto, F. M., Silva, M. J., Coutinho, P. M. (2007). Measuring semantic similarity between gene ontology terms. Data & Knowledge Engineering, 61(1), 137–152. DOI 10.1016/j.datak.2006.05.003. [Google Scholar] [CrossRef]
28. Breiman, L. (2001). Random forests. Machine Learning, 45(1), 5–32. DOI 10.1023/A:1010933404324. [Google Scholar] [CrossRef]
29. Chen, J., Li, Q., Wang, H., Deng, M. (2019). A machine learning ensemble approach based on random forest and radial basis function neural network for risk evaluation of regional flood disaster: A case study of the Yangtze river delta, China. International Journal of Environmental Research and Public Health, 17(1), 49. DOI 10.3390/ijerph17010049. [Google Scholar] [CrossRef]
30. Fröhlich, H., Speer, N., Poustka, A., Beissbarth, T. (2007). GOSim--An R-package for computation of information theoretic GO similarities between terms and gene products. BMC Bioinformatics, 8, 166. DOI 10.1186/1471-2105-8-166. [Google Scholar] [CrossRef]
31. Chawla, N. V., Bowyer, K. W., Hall, L. O., Kegelmeyer, W. P. (2002). SMOTE: Synthetic minority over-sampling technique. Journal of Artificial Intelligence Research, 16, 341–378. DOI 10.1613/jair.953. [Google Scholar] [CrossRef]
32. Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z., Zhang, B. (2019). Webgestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Research, 47(W1), W199–W205. DOI 10.1093/nar/gkz401. [Google Scholar] [CrossRef]
33. Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., Morishima, K. (2017). KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research, 45(D1), D353–D361. DOI 10.1093/nar/gkw1092. [Google Scholar] [CrossRef]
34. Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D. et al. (2015). STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Research, 43(Database Issue), D447–D452. DOI 10.1093/nar/gku1003. [Google Scholar] [CrossRef]
35. Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T. et al. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. DOI 10.1101/gr.1239303. [Google Scholar] [CrossRef]
36. Bader, G. D., Hogue, C. W. (2003). An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics, 4(1), 1–27. DOI 10.1186/1471-2105-4-1. [Google Scholar] [CrossRef]
37. Koff, W. C., Williams, M. A. (2020). COVID-19 and immunity in aging populations—A new research agenda. New England Journal of Medicine, 383(9), 804–805. DOI 10.1056/NEJMp2006761. [Google Scholar] [CrossRef]
38. Maremanda, K. P., Sundar, I. K., Li, D., Rahman, I. (2020). Age-dependent assessment of genes involved in cellular senescence, telomere, and mitochondrial pathways in human lung tissue of smokers, COPD, and IPF: Associations with SARS-CoV-2 COVID-19 ACE2-tMPRSS2-furin-dPP4 axis. Frontiers in Pharmacology, 11, 1356. DOI 10.3389/fphar.2020.584637. [Google Scholar] [CrossRef]
39. Samuel, C. E. (2001). Antiviral actions of interferons. Clinical Microbiology Reviews, 14(4), 778–809. DOI 10.1128/CMR.14.4.778-809.2001. [Google Scholar] [CrossRef]
40. Prasad, K., Khatoon, F., Rashid, S., Ali, N., AlAsmari, A. F. et al. (2020). Targeting hub genes and pathways of innate immune response in COVID-19: A network biology perspective. International Journal of Biological Macromolecules, 163, 1–8. DOI 10.1016/j.ijbiomac.2020.06.228. [Google Scholar] [CrossRef]
41. Shaath, H., Vishnubalaji, R., Elkord, E., Alajez, N. M. (2020). Single-cell transcriptome analysis highlights a role for neutrophils and inflammatory macrophages in the pathogenesis of severe COVID-19. Cells, 9(11), 2374. DOI 10.3390/cells9112374. [Google Scholar] [CrossRef]
42. de Wit, E., van Doremalen, N., Falzarano, D., Munster, V. J. (2016). SARS and MERS: Recent insights into emerging coronaviruses. Nature Reviews Microbiology, 14(8), 523–534. DOI 10.1038/nrmicro.2016.81. [Google Scholar] [CrossRef]
43. Shi, C. S., Qi, H. Y., Boularan, C., Huang, N. N., Abu-Asab, M. et al. (2014). SARS-Coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. The Journal of Immunology, 193(6), 3080–3089. DOI 10.4049/jimmunol.1303196. [Google Scholar] [CrossRef]
44. Liu, G., Lee, J. H., Parker, Z. M., Acharya, D., Chiang, J. J. et al. (2021). ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nature Microbiology, 6(4), 467–478. DOI 10.1038/s41564-021-00884-1. [Google Scholar] [CrossRef]
 | This work is licensed under a Creative Commons Attribution 4.0 International License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |